

Moving toward a system genetics view of disease
- PMID: 17653589
- PMCID: PMC1998874
- DOI: 10.1007/s00335-007-9040-6
Moving toward a system genetics view of disease
Abstract
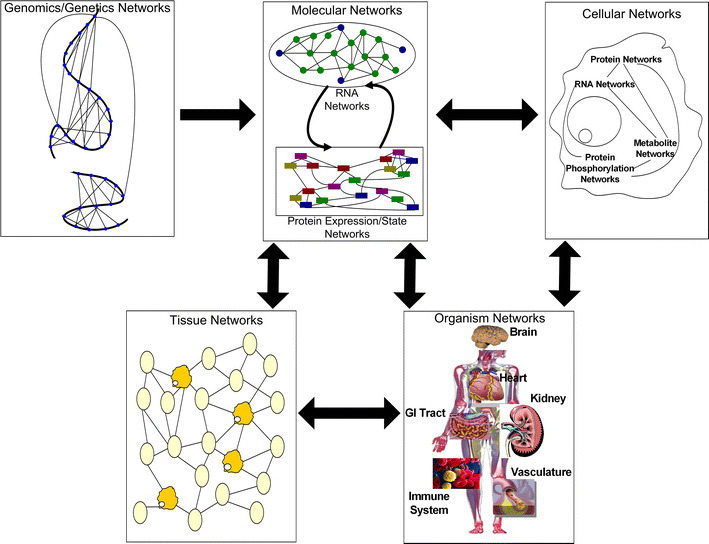
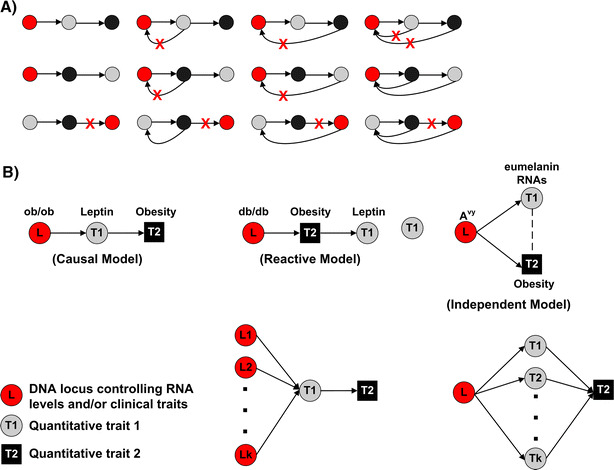
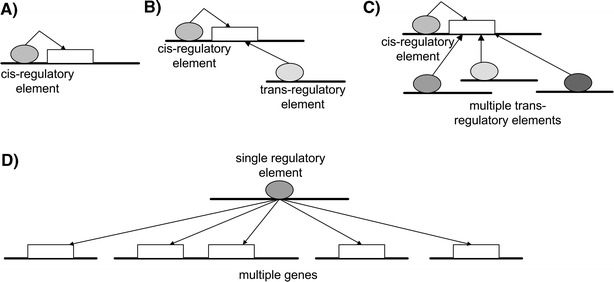
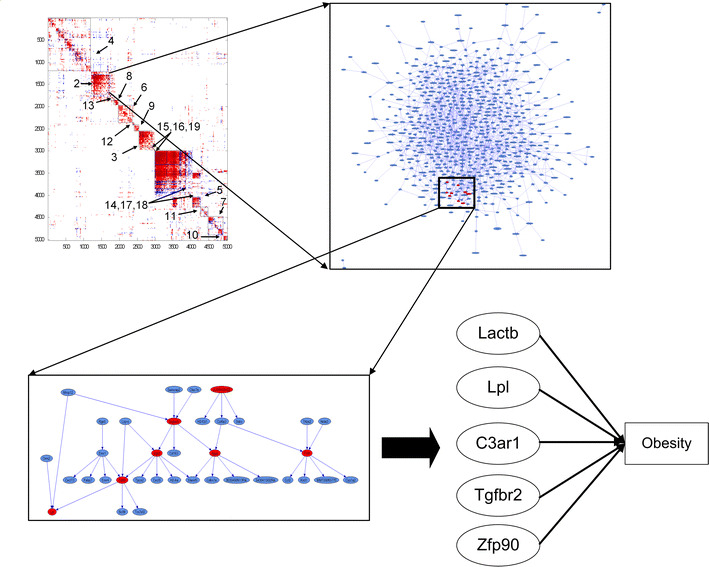
Testing hundreds of thousands of DNA markers in human, mouse, and other species for association to complex traits like disease is now a reality. However, information on how variations in DNA impact complex physiologic processes flows through transcriptional and other molecular networks. In other words, DNA variations impact complex diseases through the perturbations they cause to transcriptional and other biological networks, and these molecular phenotypes are intermediate to clinically defined disease. Because it is also now possible to monitor transcript levels in a comprehensive fashion, integrating DNA variation, transcription, and phenotypic data has the potential to enhance identification of the associations between DNA variation and diseases like obesity and diabetes, as well as characterize those parts of the molecular networks that drive these diseases. Toward that end, we review methods for integrating expression quantitative trait loci (eQTLs), gene expression, and clinical data to infer causal relationships among gene expression traits and between expression and clinical traits. We further describe methods to integrate these data in a more comprehensive manner by constructing coexpression gene networks that leverage pairwise gene interaction data to represent more general relationships. To infer gene networks that capture causal information, we describe a Bayesian algorithm that further integrates eQTLs, expression, and clinical phenotype data to reconstruct whole-gene networks capable of representing causal relationships among genes and traits in the network. These emerging network approaches, aimed at processing high-dimensional biological data by integrating data from multiple sources, represent some of the first steps in statistical genetics to identify multiple genetic perturbations that alter the states of molecular networks and that in turn push systems into disease states. Evolving statistical procedures that operate on networks will be critical to extracting information related to complex phenotypes like disease, as research goes beyond a single-gene focus. The early successes achieved with the methods described herein suggest that these more integrative genomics approaches to dissecting disease traits will significantly enhance the identification of key drivers of disease beyond what could be achieved by genetic association studies alone.
Figures

References
-
- Alberts B (2002) Molecular biology of the cell (New York: Garland Science), p xxxiv
-
- Barabasi AL, Albert R. Emergence of scaling in random networks. Science. 1999;286:509–512. - PubMed
-
- Barabasi AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5:101–113. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
