

Comparative mitochondrial genomics of snakes: extraordinary substitution rate dynamics and functionality of the duplicate control region
- PMID: 17655768
- PMCID: PMC1950710
- DOI: 10.1186/1471-2148-7-123
Comparative mitochondrial genomics of snakes: extraordinary substitution rate dynamics and functionality of the duplicate control region
Abstract
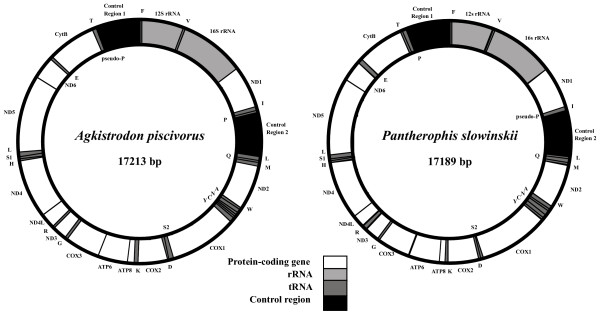
Background: The mitochondrial genomes of snakes are characterized by an overall evolutionary rate that appears to be one of the most accelerated among vertebrates. They also possess other unusual features, including short tRNAs and other genes, and a duplicated control region that has been stably maintained since it originated more than 70 million years ago. Here, we provide a detailed analysis of evolutionary dynamics in snake mitochondrial genomes to better understand the basis of these extreme characteristics, and to explore the relationship between mitochondrial genome molecular evolution, genome architecture, and molecular function. We sequenced complete mitochondrial genomes from Slowinski's corn snake (Pantherophis slowinskii) and two cottonmouths (Agkistrodon piscivorus) to complement previously existing mitochondrial genomes, and to provide an improved comparative view of how genome architecture affects molecular evolution at contrasting levels of divergence.
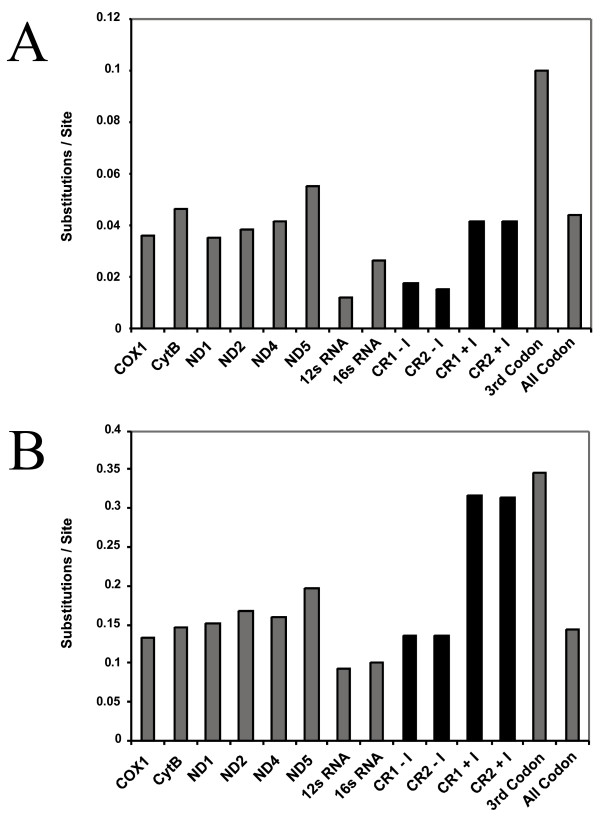
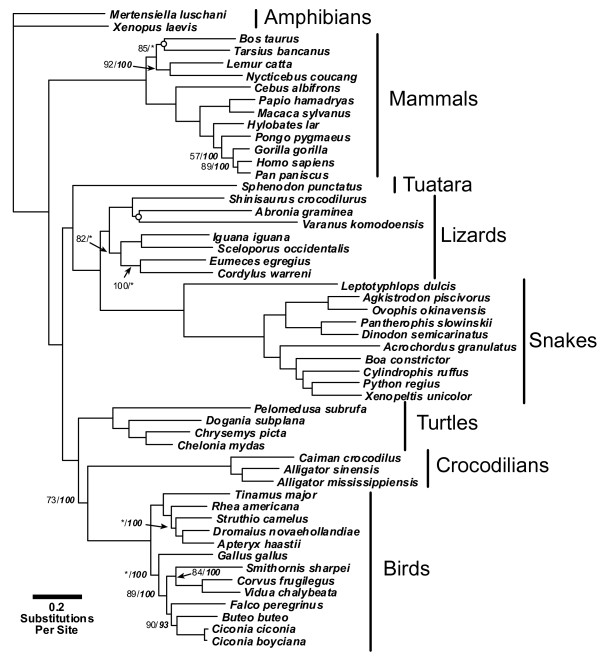
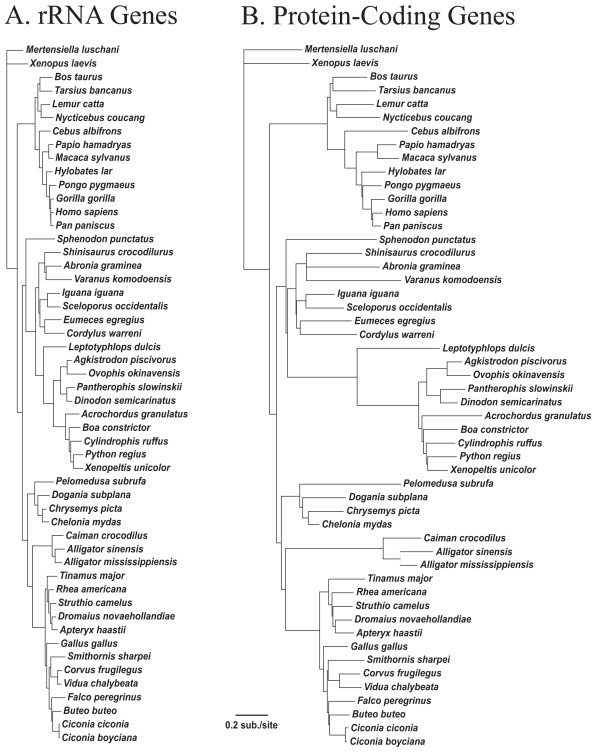
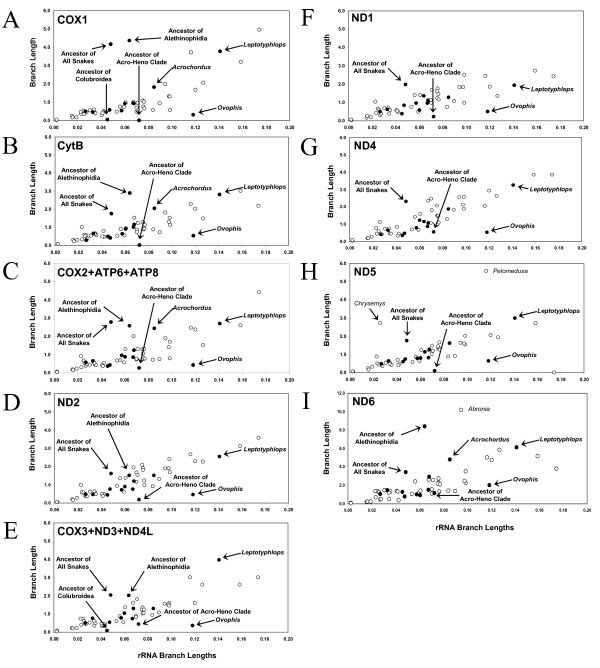
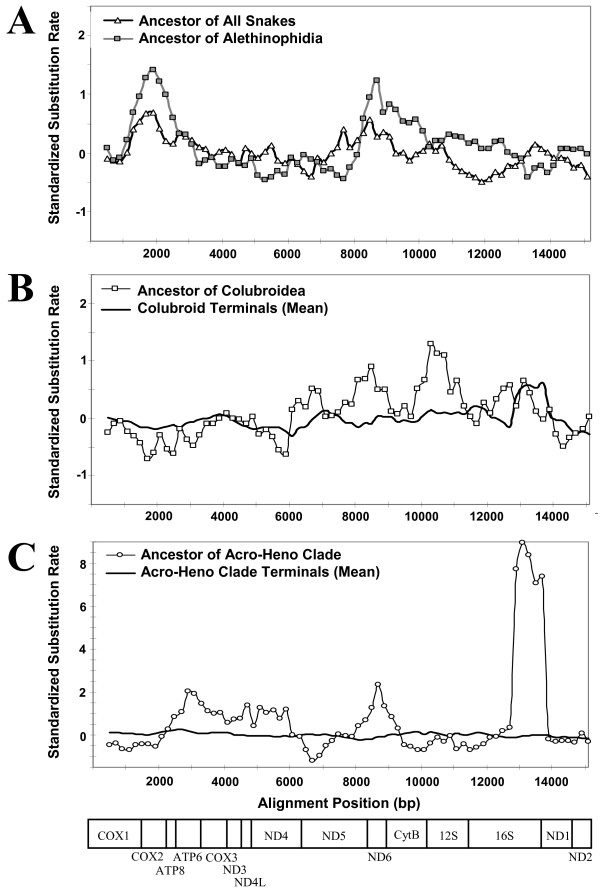
Results: We present a Bayesian genetic approach that suggests that the duplicated control region can function as an additional origin of heavy strand replication. The two control regions also appear to have different intra-specific versus inter-specific evolutionary dynamics that may be associated with complex modes of concerted evolution. We find that different genomic regions have experienced substantial accelerated evolution along early branches in snakes, with different genes having experienced dramatic accelerations along specific branches. Some of these accelerations appear to coincide with, or subsequent to, the shortening of various mitochondrial genes and the duplication of the control region and flanking tRNAs.
Conclusion: Fluctuations in the strength and pattern of selection during snake evolution have had widely varying gene-specific effects on substitution rates, and these rate accelerations may have been functionally related to unusual changes in genomic architecture. The among-lineage and among-gene variation in rate dynamics observed in snakes is the most extreme thus far observed in animal genomes, and provides an important study system for further evaluating the biochemical and physiological basis of evolutionary pressures in vertebrate mitochondria.
Figures

References
-
- Reyes A, Gissi C, Pesole G, Saccone C. Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol Biol Evol. 1998;15:957–966. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
