

Toward rational protein crystallization: A Web server for the design of crystallizable protein variants
- PMID: 17656576
- PMCID: PMC2203352
- DOI: 10.1110/ps.072914007
Toward rational protein crystallization: A Web server for the design of crystallizable protein variants
Abstract
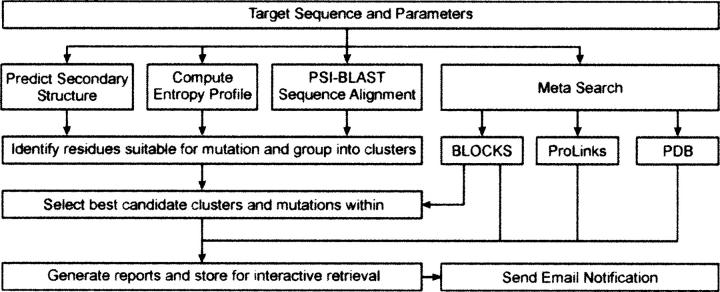
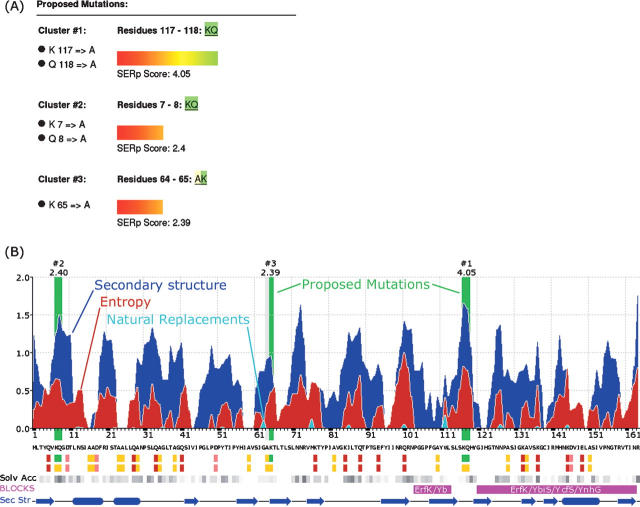
Growing well-diffracting crystals constitutes a serious bottleneck in structural biology. A recently proposed crystallization methodology for "stubborn crystallizers" is to engineer surface sequence variants designed to form intermolecular contacts that could support a crystal lattice. This approach relies on the concept of surface entropy reduction (SER), i.e., the replacement of clusters of flexible, solvent-exposed residues with residues with lower conformational entropy. This strategy minimizes the loss of conformational entropy upon crystallization and renders crystallization thermodynamically favorable. The method has been successfully used to crystallize more than 15 novel proteins, all stubborn crystallizers. But the choice of suitable sites for mutagenesis is not trivial. Herein, we announce a Web server, the surface entropy reduction prediction server (SERp server), designed to identify mutations that may facilitate crystallization. Suggested mutations are predicted based on an algorithm incorporating a conformational entropy profile, a secondary structure prediction, and sequence conservation. Minor considerations include the nature of flanking residues and gaps between mutation candidates. While designed to be used with default values, the server has many user-controlled parameters allowing for considerable flexibility. Within, we discuss (1) the methodology of the server, (2) how to interpret the results, and (3) factors that must be considered when selecting mutations. We also attempt to benchmark the server by comparing the server's predictions with successful SER structures. In most cases, the structure yielding mutations were easily identified by the SERp server. The server can be accessed at http://www.doe-mbi.ucla.edu/Services/SER.
Figures
References
-
- Boeshans K.M., Liu, F., Peng, G., Idler, W., Jang, S.I., Marekov, L., Black, L., and Ahvazi, B. 2006. Purification, crystallization and preliminary X-ray diffraction analysis of the phage T4 vertex protein gp24 and its mutant forms. Protein Expr. Purif. 49: 2: 235–243. - PubMed
-
- Bowers P.M., Pellegrini, M., Fierro, J., and Eisenberg, D. 2004. Prolinks: A database of protein functional linkages derived from coevolution. Genome Biol. http://genomebiology.com/2004/5/5/R35. - PMC - PubMed
-
- Conte L.L., Chothia, C., and Janin, J. 1999. The atomic structure of protein-protein recognition sites. J. Mol. Biol. 285: 2177–2198. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
