

Excitation-contraction coupling and mitochondrial energetics
- PMID: 17657400
- PMCID: PMC2785083
- DOI: 10.1007/s00395-007-0666-z
Excitation-contraction coupling and mitochondrial energetics
Abstract
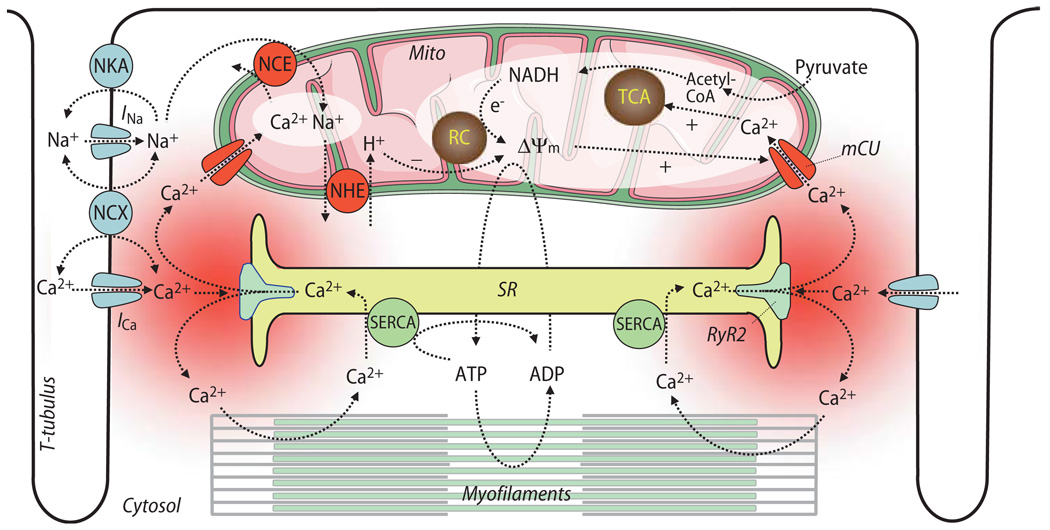
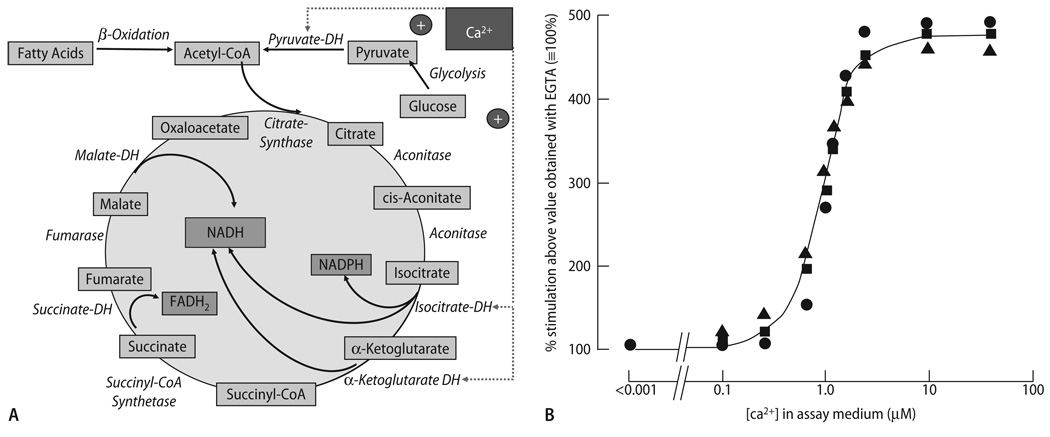
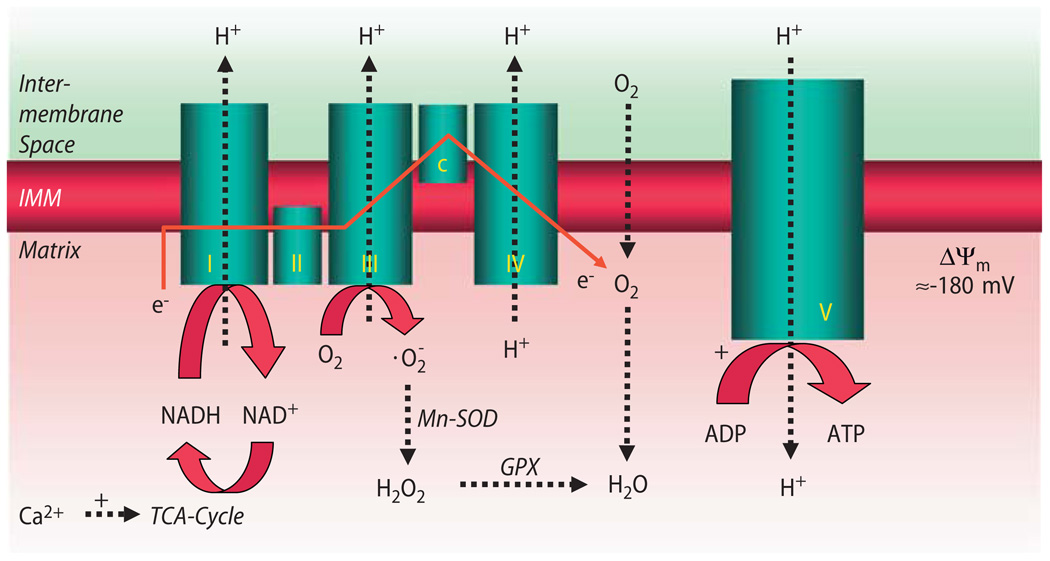
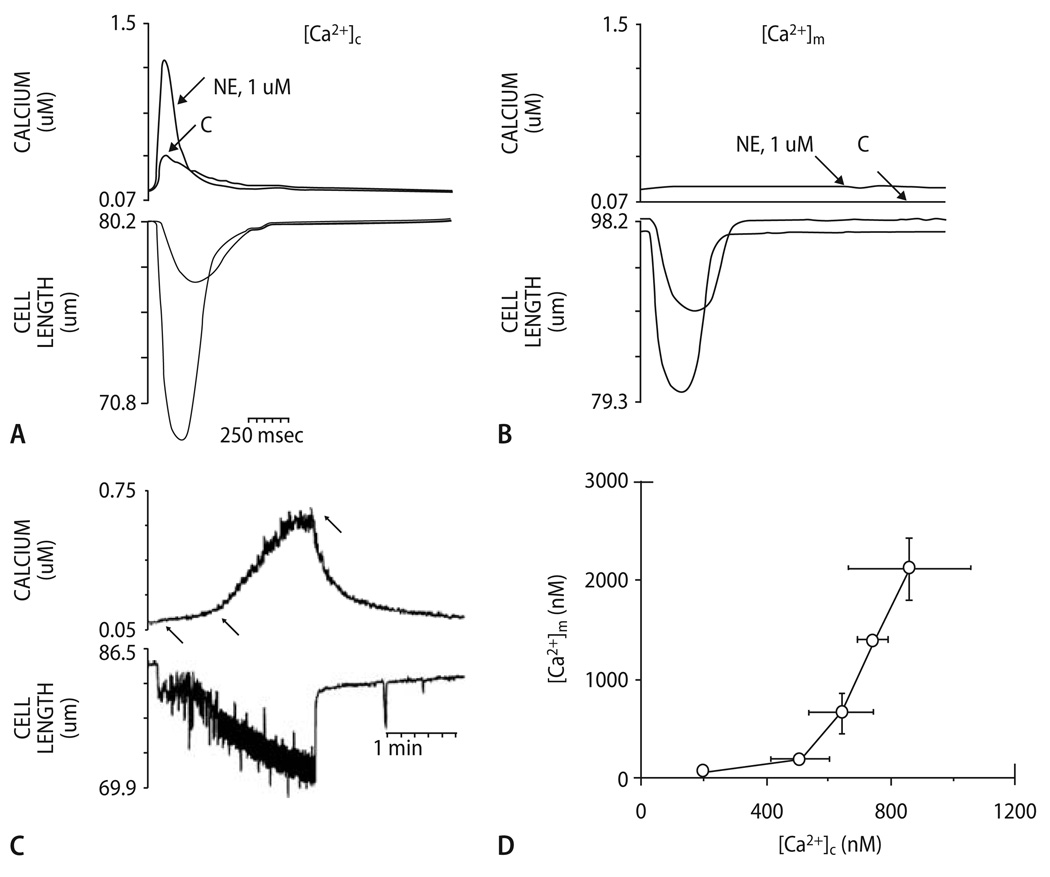
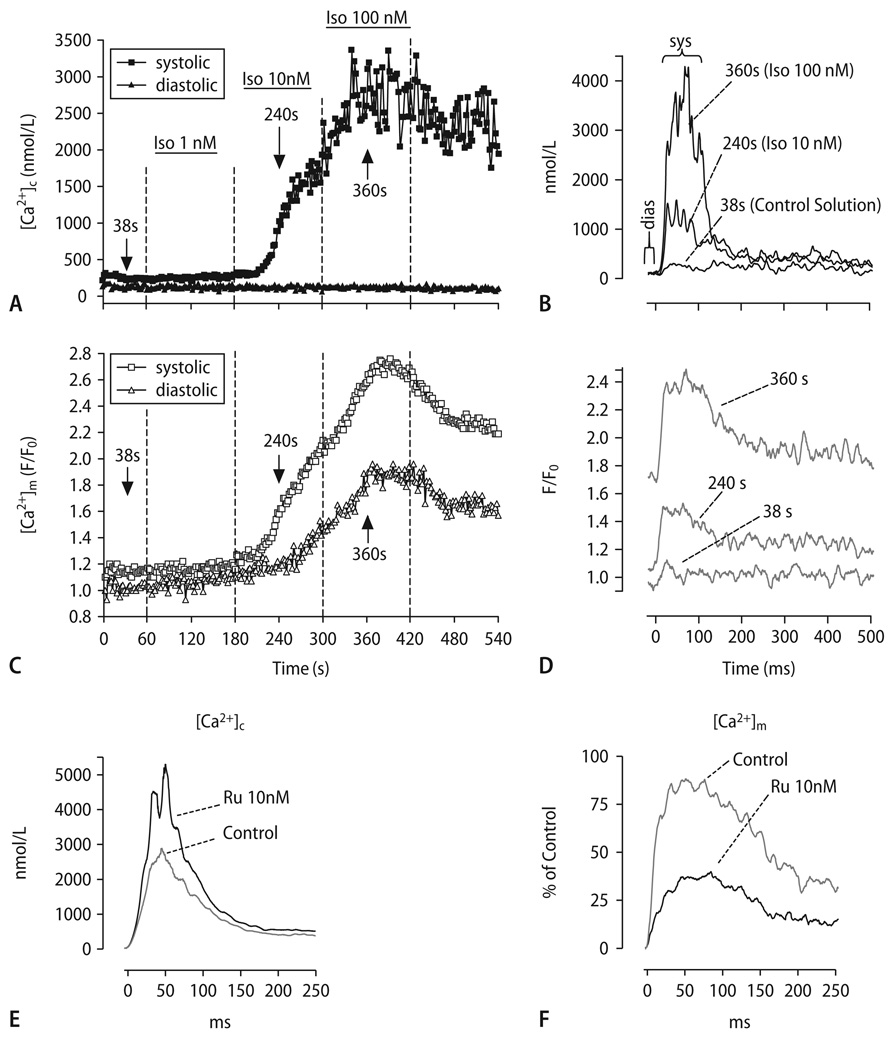
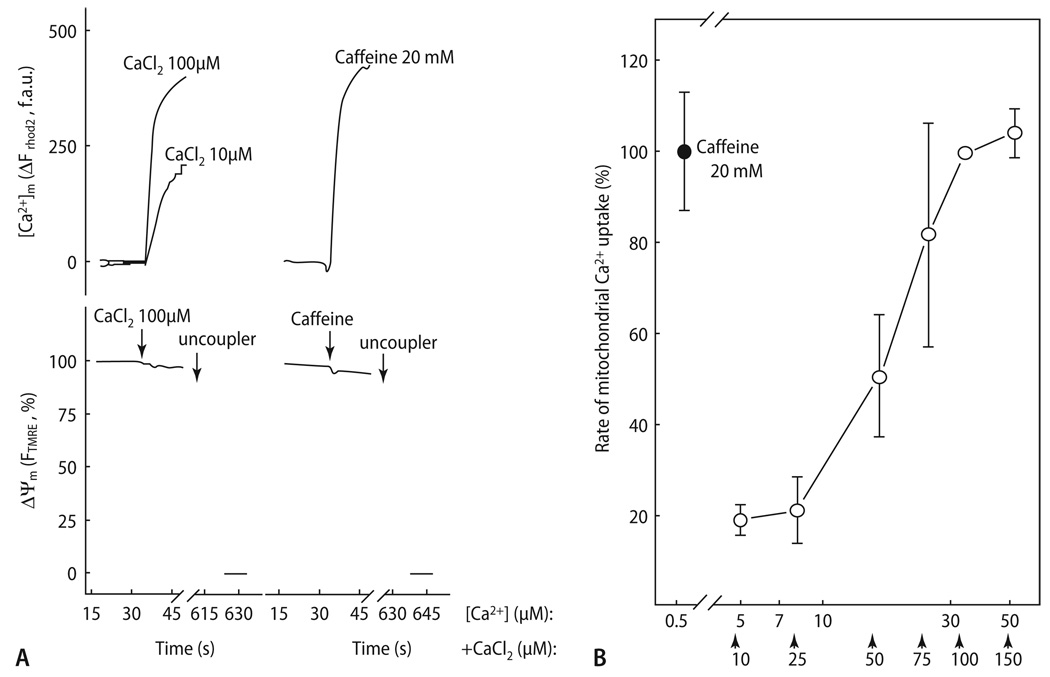
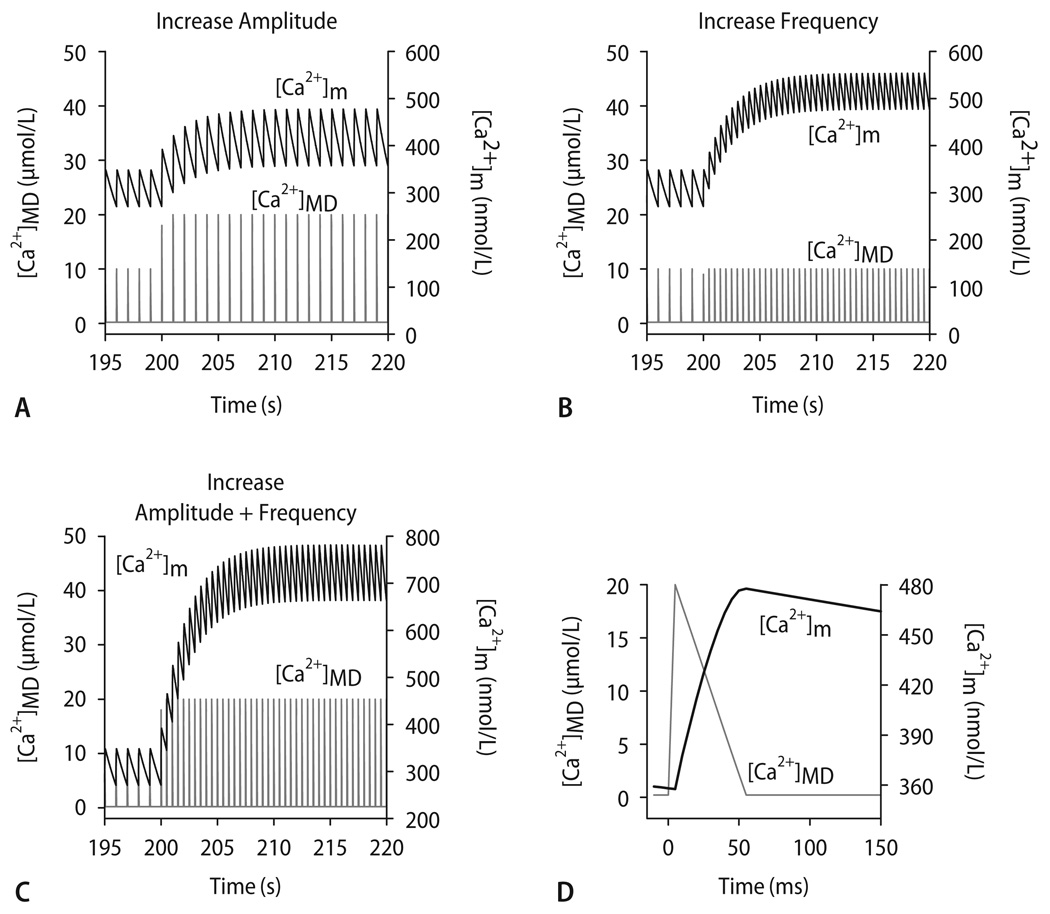
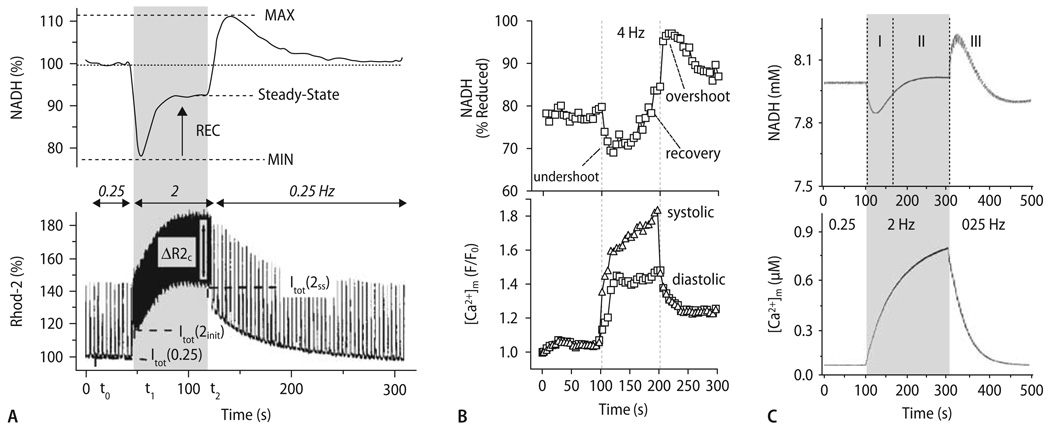
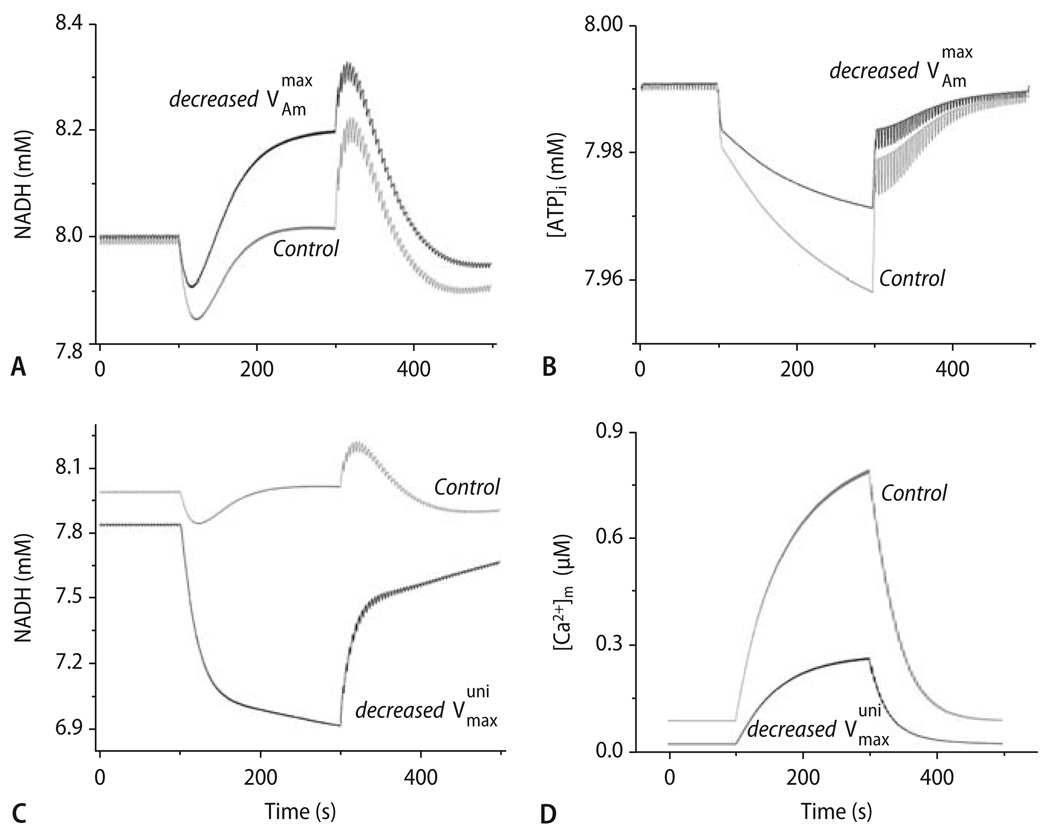
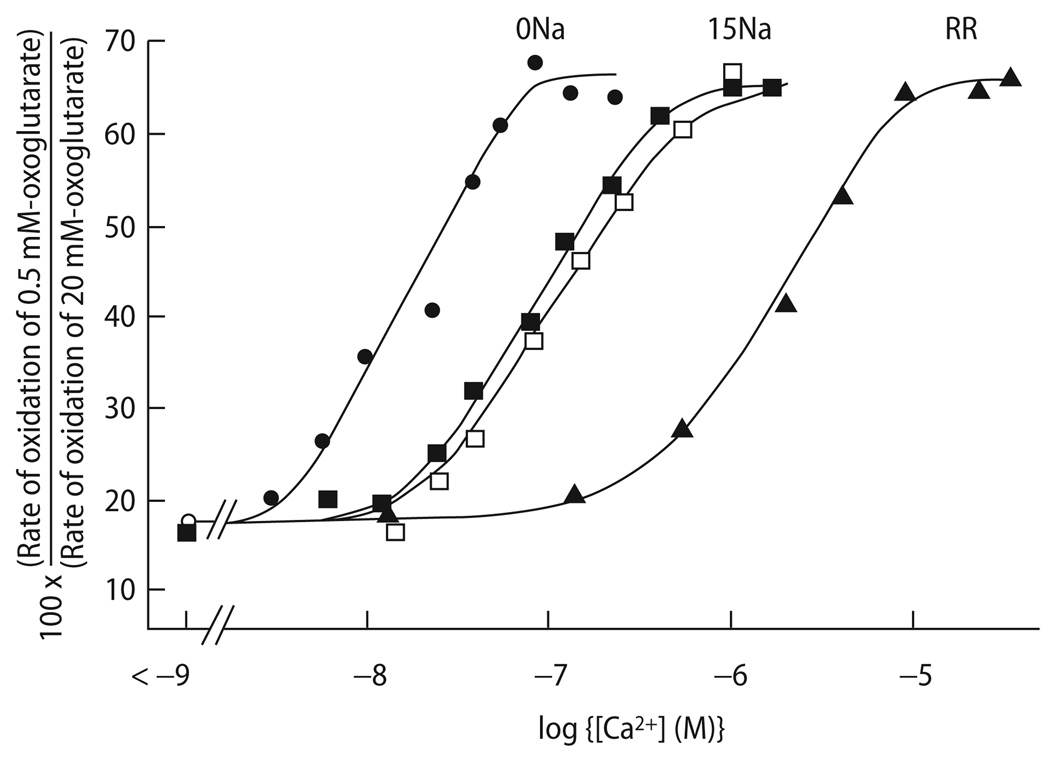
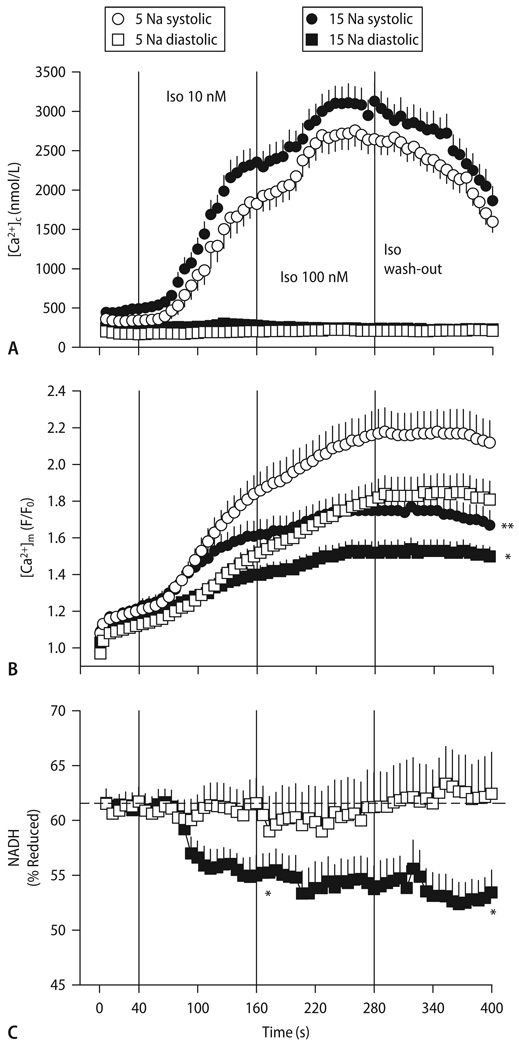
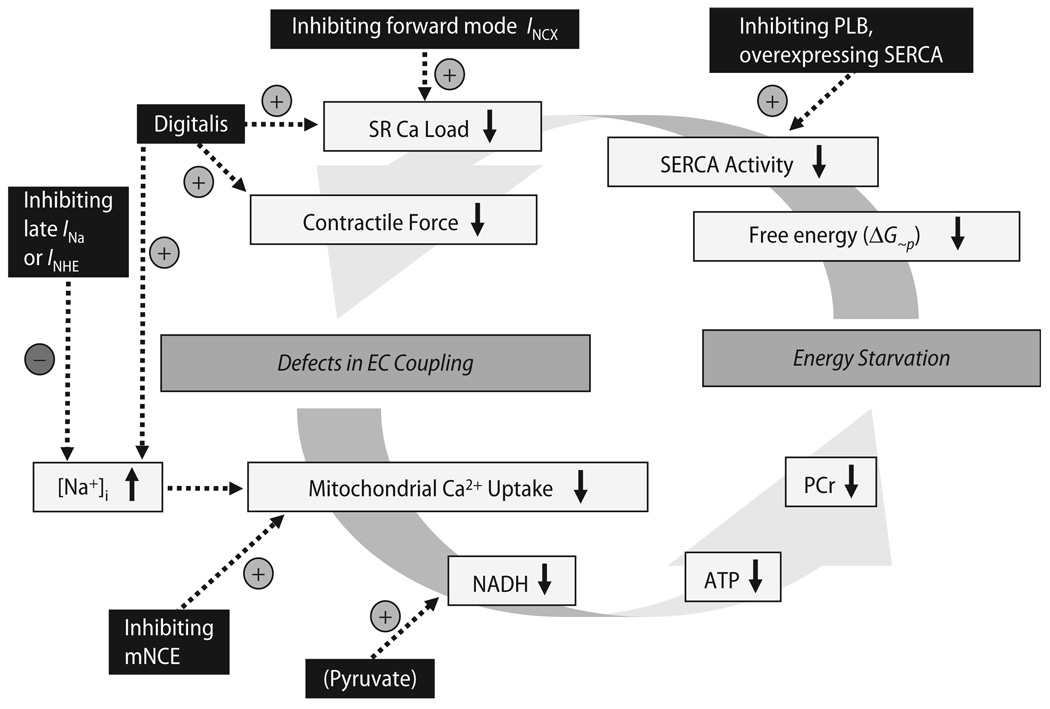
Cardiac excitation-contraction (EC) coupling consumes vast amounts of cellular energy, most of which is produced in mitochondria by oxidative phosphorylation. In order to adapt the constantly varying workload of the heart to energy supply, tight coupling mechanisms are essential to maintain cellular pools of ATP, phosphocreatine and NADH. To our current knowledge, the most important regulators of oxidative phosphorylation are ADP, Pi, and Ca2+. However, the kinetics of mitochondrial Ca2+-uptake during EC coupling are currently a matter of intense debate. Recent experimental findings suggest the existence of a mitochondrial Ca2+ microdomain in cardiac myocytes, justified by the close proximity of mitochondria to the sites of cellular Ca2+ release, i. e., the ryanodine receptors of the sarcoplasmic reticulum. Such a Ca2+ microdomain could explain seemingly controversial results on mitochondrial Ca2+ uptake kinetics in isolated mitochondria versus whole cardiac myocytes. Another important consideration is that rapid mitochondrial Ca2+ uptake facilitated by microdomains may shape cytosolic Ca2+ signals in cardiac myocytes and have an impact on energy supply and demand matching. Defects in EC coupling in chronic heart failure may adversely affect mitochondrial Ca2+ uptake and energetics, initiating a vicious cycle of contractile dysfunction and energy depletion. Future therapeutic approaches in the treatment of heart failure could be aimed at interrupting this vicious cycle.
Figures

References
-
- Abozguia K, Clarke K, Lee L, Frenneaux M. Modification of myocardial substrate use as a therapy for heart failure. Nat Clin Pract Cardiovasc Med. 2006;3:490–498. - PubMed
-
- Aker S, Snabaitis AK, Konietzka I, Van De Sand A, Bongler K, Avkiran M, Heusch G, Schulz R. Inhibition of the Na+/H+ exchanger attenuates the deterioration of ventricular function during pacing-induced heart failure in rabbits. Cardiovasc Res. 2004;63:273–282. - PubMed
-
- Auffermann W, Wu ST, Parmley WW, Wikman-Coffelt J. Glycolysis in heart failure: a 31P-NMR and surface fluorometry study. Basic Res Cardiol. 1990;85:342–357. - PubMed
-
- Baartscheer A, Schumacher CA, van Borren MM, Belterman CN, Coronel R, Fiolet JW. Increased Na+/H+-exchange activity is the cause of increased [Na+]i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc Res. 2003;57:1015–1024. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
