

Toward a systems biology of mouse inner ear organogenesis: gene expression pathways, patterns and network analysis
- PMID: 17660535
- PMCID: PMC2013721
- DOI: 10.1534/genetics.107.078584
Toward a systems biology of mouse inner ear organogenesis: gene expression pathways, patterns and network analysis
Abstract
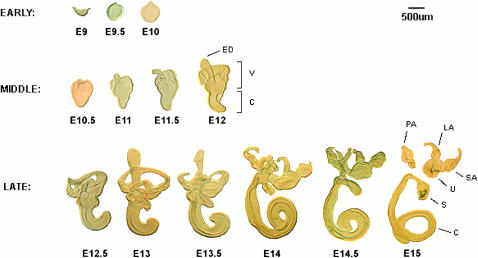
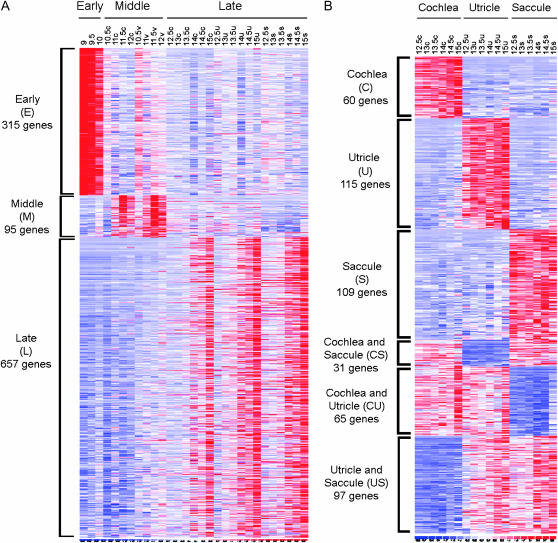
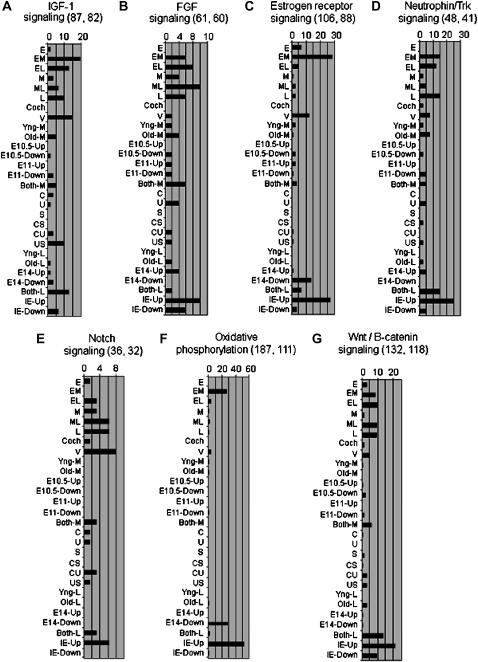
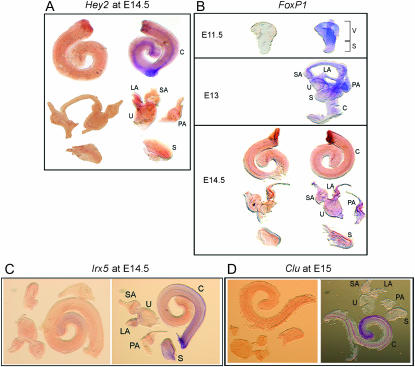
We describe the most comprehensive study to date on gene expression during mouse inner ear (IE) organogenesis. Samples were microdissected from mouse embryos at E9-E15 in half-day intervals, a period that spans all of IE organogenesis. These included separate dissections of all discernible IE substructures such as the cochlea, utricle, and saccule. All samples were analyzed on high density expression microarrays under strict statistical filters. Extensive confirmatory tests were performed, including RNA in situ hybridizations. More than 5000 genes significantly varied in expression according to developmental stage, tissue, or both and defined 28 distinct expression patterns. For example, upregulation of 315 genes provided a clear-cut "signature" of early events in IE specification. Additional, clear-cut, gene expression signatures marked specific structures such as the cochlea, utricle, or saccule throughout late IE development. Pathway analysis identified 53 signaling cascades enriched within the 28 patterns. Many novel pathways, not previously implicated in IE development, including beta-adrenergic, amyloid, estrogen receptor, circadian rhythm, and immune system pathways, were identified. Finally, we identified positional candidate genes in 54 uncloned nonsyndromic human deafness intervals. This detailed analysis provides many new insights into the spatial and temporal genetic specification of this complex organ system.
Figures




Similar articles
-
Analysis of genes from inner ear developmental-stage cDNA subtraction reveals molecular regionalization of the otic capsule.Dev Biol. 2004 Apr 1;268(1):7-23. doi: 10.1016/j.ydbio.2003.11.023. Dev Biol. 2004. PMID: 15031101
-
Tectorin mRNA expression is spatially and temporally restricted during mouse inner ear development.J Comp Neurol. 1999 Mar 8;405(2):271-80. J Comp Neurol. 1999. PMID: 10023815
-
A comprehensive catalogue of the coding and non-coding transcripts of the human inner ear.Hear Res. 2016 Mar;333:266-274. doi: 10.1016/j.heares.2015.08.013. Epub 2015 Sep 1. Hear Res. 2016. PMID: 26341477 Free PMC article.
-
Understanding inner ear development with gene expression profiling.J Neurobiol. 2002 Nov 5;53(2):276-85. doi: 10.1002/neu.10125. J Neurobiol. 2002. PMID: 12382281 Review.
-
Information for gene networks in inner ear development: a study centered on the transcription factor gata2.Hear Res. 2007 May;227(1-2):32-40. doi: 10.1016/j.heares.2006.04.006. Epub 2006 Jun 23. Hear Res. 2007. PMID: 16797894 Review.
Cited by
-
The applications of single-cell genomics.Hum Mol Genet. 2013 Oct 15;22(R1):R22-6. doi: 10.1093/hmg/ddt377. Epub 2013 Aug 6. Hum Mol Genet. 2013. PMID: 23922233 Free PMC article. Review.
-
Identification and characterization of mouse otic sensory lineage genes.Front Cell Neurosci. 2015 Mar 19;9:79. doi: 10.3389/fncel.2015.00079. eCollection 2015. Front Cell Neurosci. 2015. PMID: 25852475 Free PMC article.
-
Enriched Differentiation of Human Otic Sensory Progenitor Cells Derived From Induced Pluripotent Stem Cells.Front Mol Neurosci. 2018 Dec 20;11:452. doi: 10.3389/fnmol.2018.00452. eCollection 2018. Front Mol Neurosci. 2018. PMID: 30618604 Free PMC article.
-
A symphony of inner ear developmental control genes.BMC Genet. 2010 Jul 16;11:68. doi: 10.1186/1471-2156-11-68. BMC Genet. 2010. PMID: 20637105 Free PMC article. Review.
-
[Regenerative medicine in the treatment of sensorineural hearing loss].HNO. 2008 Mar;56(3):288-300. doi: 10.1007/s00106-008-1689-y. HNO. 2008. PMID: 18288464 Review. German.
References
-
- Anagnostopoulos, A. V., 2002. A compendium of mouse knockouts with inner ear defects. Trends Genet. 18: 499. - PubMed
-
- Anniko, M., 1983. Cytodifferentiation of cochlear hair cells. Am. J. Otolaryngol. 4: 375–388. - PubMed
-
- Avraham, K. B, 2003. Mouse models for deafness: lessons for the human inner ear and hearing loss. Ear Hear. 24: 332–341. - PubMed
-
- Beisel, K. W., T. Shiraki, K. A. Morris, C. Pompeia, B. Kachar et al., 2004. Identification of unique transcripts from a mouse full-length, subtracted inner ear cDNA library. Genomics 83: 1012–1023. - PubMed
-
- Bhave, S. A., E. C. Oesterle and M. D. Coltrera, 1998. Macrophage and microglia-like cells in the avian inner ear. J. Comp. Neurol. 398: 241–256. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
