

Induction of tolerance to human arylsulfatase A in a mouse model of metachromatic leukodystrophy
- PMID: 17660863
- PMCID: PMC1933260
- DOI: 10.2119/2007-00063.Matzner
Induction of tolerance to human arylsulfatase A in a mouse model of metachromatic leukodystrophy
Abstract
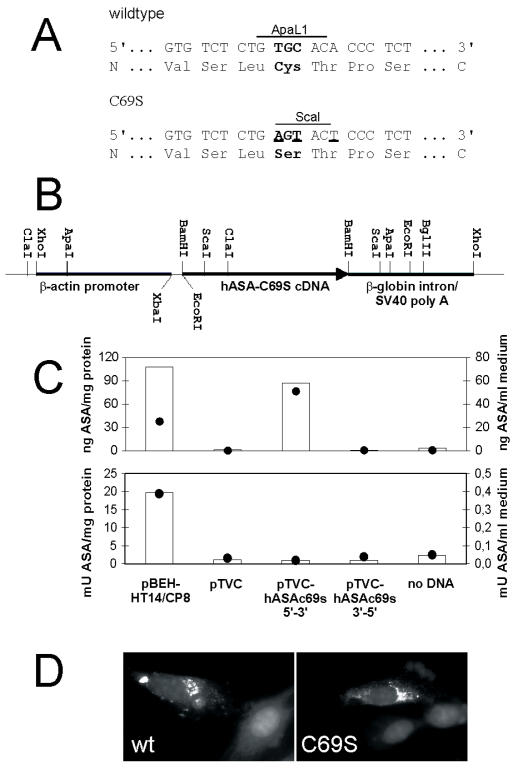
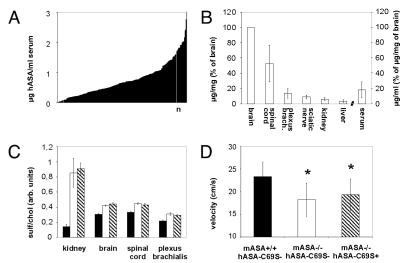
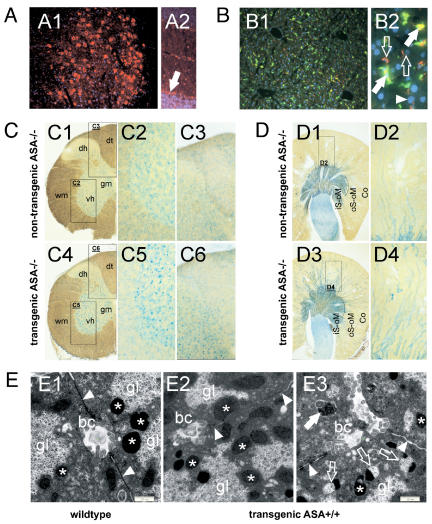
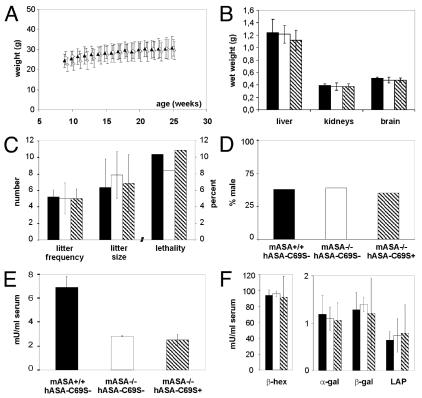
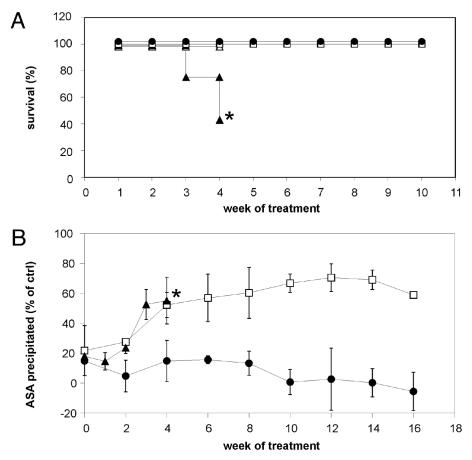
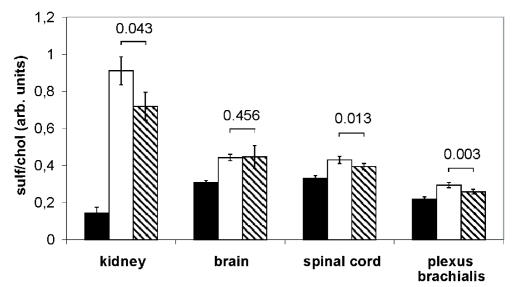
A deficiency of arylsulfatase A (ASA) causes metachromatic leukodystrophy (MLD), a lysosomal storage disorder characterized by accumulation of sulfatide, a severe neurological phenotype and early death. The efficacy of enzyme replacement therapy (ERT) has previously been determined in ASA knockout (ASA-/-) mice representing the only available animal model for MLD. Repeated intravenous injection of human ASA (hASA) improved the nervous system pathology and function, but also elicited a progressive humoral immune response leading to treatment resistance, anaphylactic reactions, and high mortality. In contrast to ASA-/- mice, most MLD patients express mutant hASA which may entail immunological tolerance to substituted wildtype hASA and thus protect from immunological complications. To test this notion, a cysteine-to-serine substitution was introduced into the active site of the hASA and the resulting inactive hASA-C69S variant was constitutively expressed in ASA-/- mice. Mice with sub-to supranormal levels of mutant hASA expression were analyzed. All mice, including those showing transgene expression below the limit of detection, were immunologically unresponsive to injected hASA. More than 100-fold overexpression did not induce an overt new phenotype except occasional intralysosomal deposition of minor amounts of glycogen in hepatocytes. Furthermore, long-term, low-dose ERT reduced sulfatide storage in peripheral tissues and the central nervous system indicating that high levels of extracellular mutant hASA do not prevent cellular uptake and lysosomal targeting of substituted wildtype hASA. Due to the tolerance to hASA and maintenance of the MLD-like phenotype, the novel transgenic strain may be particularly advantageous to assess the benefit and risk of long-term ERT.
Figures
Similar articles
-
Enzyme replacement improves nervous system pathology and function in a mouse model for metachromatic leukodystrophy.Hum Mol Genet. 2005 May 1;14(9):1139-52. doi: 10.1093/hmg/ddi126. Epub 2005 Mar 16. Hum Mol Genet. 2005. PMID: 15772092
-
Engineered arylsulfatase A with increased activity, stability and brain delivery for therapy of metachromatic leukodystrophy.Mol Ther. 2023 Oct 4;31(10):2962-2974. doi: 10.1016/j.ymthe.2023.08.019. Epub 2023 Aug 29. Mol Ther. 2023. PMID: 37644722 Free PMC article.
-
Intracerebroventricular enzyme infusion corrects central nervous system pathology and dysfunction in a mouse model of metachromatic leukodystrophy.Hum Mol Genet. 2011 Jul 15;20(14):2760-9. doi: 10.1093/hmg/ddr175. Epub 2011 Apr 22. Hum Mol Genet. 2011. PMID: 21515587
-
Developing therapeutic approaches for metachromatic leukodystrophy.Drug Des Devel Ther. 2013 Aug 8;7:729-45. doi: 10.2147/DDDT.S15467. eCollection 2013. Drug Des Devel Ther. 2013. PMID: 23966770 Free PMC article. Review.
-
Metachromatic leukodystrophy: molecular genetics and an animal model.J Inherit Metab Dis. 1998 Aug;21(5):564-74. doi: 10.1023/a:1005471106088. J Inherit Metab Dis. 1998. PMID: 9728336 Review.
Cited by
-
Nonclinical comparability studies of recombinant human arylsulfatase A addressing manufacturing process changes.PLoS One. 2018 Apr 19;13(4):e0195186. doi: 10.1371/journal.pone.0195186. eCollection 2018. PLoS One. 2018. PMID: 29672630 Free PMC article.
-
Dose-response evaluation of intravenous gene therapy in a symptomatic mouse model of metachromatic leukodystrophy.Mol Ther Methods Clin Dev. 2024 Apr 6;32(2):101248. doi: 10.1016/j.omtm.2024.101248. eCollection 2024 Jun 13. Mol Ther Methods Clin Dev. 2024. PMID: 38680552 Free PMC article.
-
Deep proteomic profiling unveils arylsulfatase A as a non-alcoholic steatohepatitis inducible hepatokine and regulator of glycemic control.Nat Commun. 2022 Mar 10;13(1):1259. doi: 10.1038/s41467-022-28889-2. Nat Commun. 2022. PMID: 35273160 Free PMC article.
-
Cross-species efficacy of AAV-mediated ARSA replacement for metachromatic leukodystrophy.J Clin Invest. 2025 Jun 19;135(16):e185001. doi: 10.1172/JCI185001. eCollection 2025 Aug 15. J Clin Invest. 2025. PMID: 40536808 Free PMC article.
-
Quantification of sulfatides and lysosulfatides in tissues and body fluids by liquid chromatography-tandem mass spectrometry.J Lipid Res. 2015 Apr;56(4):936-43. doi: 10.1194/jlr.M057232. Epub 2015 Jan 27. J Lipid Res. 2015. PMID: 25632048 Free PMC article.
References
-
- von Figura K, Gieselmann V, Jaeken J. Metachromatic leukodystrophy. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. Mc Graw-Hill; New York, USA: 2001. pp. 3695–724.
-
- Sevin C, Aubourg P, Cartier N. Enzyme, cell and gene-based therapies for metachromatic leukodystrophy. J Inherit Metab Dis. 2007;30:175–83. - PubMed
-
- Polten A, Fluharty AL, Fluharty CB, Kappler J, von Figura K, Gieselmann V. Molecular basis of different forms of metachromatic leukodystrophy. N Engl J Med. 1991;324:18–22. - PubMed
-
- Kyewski B, Klein L. A central role for central tolerance. Annu Rev Immunol. 2006;24:571–606. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical