

Further evidence for genetic heterogeneity of distal HMN type V, CMT2 with predominant hand involvement and Silver syndrome
- PMID: 17663003
- PMCID: PMC3272403
- DOI: 10.1016/j.jns.2007.06.047
Further evidence for genetic heterogeneity of distal HMN type V, CMT2 with predominant hand involvement and Silver syndrome
Abstract
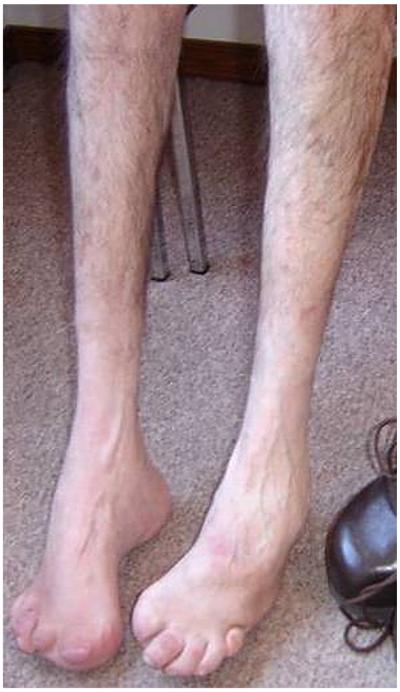
Objective: Distal hereditary motor neuropathy type V (dHMN-V) and Charcot-Marie-Tooth syndrome (CMT) type 2 presenting with predominant hand involvement, also known as CMT2D and Silver syndrome (SS) are rare phenotypically overlapping diseases which can be caused by mutations in the Berardinelli-Seip Congenital Lipodystrophy 2 (BSCL2) and in the glycyl-tRNA synthetase encoding (GARS) genes. Mutations in the heat-shock proteins HSPB1 and HSPB8 can cause related distal hereditary motor neuropathies (dHMN) and are considered candidates for dHMN-V, CMT2, and SS.
Design: To define the frequency and distribution of mutations in the GARS, BSCL2, HSPB1 and HSPB8 genes we screened 33 unrelated sporadic and familial patients diagnosed as either dHMN-V, CMT2D or SS. Exon 3 of the BSCL2 gene was screened in further 69 individuals with an unclassified dHMN phenotype or diagnosed as hereditary spastic paraplegia (HSP) complicated by pure motor neuropathy.
Results: Four patients diagnosed with dHMN-V or SS carried known heterozygous BSCL2 mutations (N88S and S90L). In one dHMN-V patient we detected a putative GARS mutation (A57V). No mutations were detected in HSPB1 and HSPB8. The diagnostic yield gained in the series of 33 probands was 12% for BSCL2 mutations and 3% for GARS mutations. In the series of unclassified dHMN and complicated HSP cases no mutations were found.
Conclusions: Our data confirm that most likely only two mutations (N88S, S90L) in exon 3 of BSCL2 may lead to dHMN-V or SS phenotypes. Mutations in GARS, HSPB1 and HSPB8. are not a common cause of dHMN-V, SS and CMT2D. We would therefore suggest that a genetic testing of dHMN-V and SS patients should begin with screening of exon 3 of the BSCL2 gene. Screening of the GARS gene is useful in patients with CMT2 with predominant hand involvement and dHMN-V. The rather low frequencies of BSCL2, GARS, HSPB1 and HSPB8 mutations in dHMN-V, CMT2D and SS patients strongly point to further genetic heterogeneity of these related disorders.
Figures
Similar articles
-
Relative contribution of mutations in genes for autosomal dominant distal hereditary motor neuropathies: a genotype-phenotype correlation study.Brain. 2008 May;131(Pt 5):1217-27. doi: 10.1093/brain/awn029. Epub 2008 Mar 5. Brain. 2008. PMID: 18325928
-
HSPB1 and HSPB8 in inherited neuropathies: study of an Italian cohort of dHMN and CMT2 patients.J Peripher Nerv Syst. 2011 Dec;16(4):287-94. doi: 10.1111/j.1529-8027.2011.00361.x. J Peripher Nerv Syst. 2011. PMID: 22176143
-
The distal hereditary motor neuropathies.J Neurol Neurosurg Psychiatry. 2012 Jan;83(1):6-14. doi: 10.1136/jnnp-2011-300952. Epub 2011 Oct 25. J Neurol Neurosurg Psychiatry. 2012. PMID: 22028385 Review.
-
Seipin S90L mutation in an Italian family with CMT2/dHMN and pyramidal signs.Muscle Nerve. 2010 Sep;42(3):448-51. doi: 10.1002/mus.21734. Muscle Nerve. 2010. PMID: 20806400
-
Distal hereditary motor neuropathies.Rev Neurol (Paris). 2024 Dec;180(10):1031-1036. doi: 10.1016/j.neurol.2023.09.005. Epub 2024 May 3. Rev Neurol (Paris). 2024. PMID: 38702287 Review.
Cited by
-
Two Novel De Novo GARS Mutations Cause Early-Onset Axonal Charcot-Marie-Tooth Disease.PLoS One. 2015 Aug 5;10(8):e0133423. doi: 10.1371/journal.pone.0133423. eCollection 2015. PLoS One. 2015. PMID: 26244500 Free PMC article.
-
Compound heterozygous mutations in glycyl-tRNA synthetase (GARS) cause mitochondrial respiratory chain dysfunction.PLoS One. 2017 Jun 8;12(6):e0178125. doi: 10.1371/journal.pone.0178125. eCollection 2017. PLoS One. 2017. PMID: 28594869 Free PMC article.
-
Exome sequencing identifies a REEP1 mutation involved in distal hereditary motor neuropathy type V.Am J Hum Genet. 2012 Jul 13;91(1):139-45. doi: 10.1016/j.ajhg.2012.05.007. Epub 2012 Jun 14. Am J Hum Genet. 2012. PMID: 22703882 Free PMC article.
-
A De Novo BSCL2 Gene S90L Mutation in a Progressive Tetraparesis with Urinary Dysfunction and Corpus Callosum Involvement.J Pediatr Genet. 2021 Sep;10(3):253-258. doi: 10.1055/s-0040-1713768. Epub 2020 Jul 8. J Pediatr Genet. 2021. PMID: 34504732 Free PMC article.
-
tRNA overexpression rescues peripheral neuropathy caused by mutations in tRNA synthetase.Science. 2021 Sep 3;373(6559):1161-1166. doi: 10.1126/science.abb3356. Epub 2021 Sep 1. Science. 2021. PMID: 34516840 Free PMC article.
References
-
- Auer-Grumbach M, Loscher WN, Wagner K, et al. Phenotypic and genotypic heterogeneity in hereditary motor neuronopathy type V: a clinical, electrophysiological and genetic study. Brain. 2000;123(Pt8):1612–23. - PubMed
-
- Auer-Grumbach M, Schlotter-Weigel B, Lochmuller H, et al. Phenotypes of the N88S Berardinelli–Seip congenital lipodystrophy 2 mutation. Ann Neurol. 2005;57:415–24. - PubMed
-
- Ionasescu V, Searby C, Sheffield VC, Roklina T, Nishimura D, Ionasescu R. Autosomal dominant Charcot–Marie–Tooth axonal neuropathy mapped on chromosome 7p (CMT2D) Hum Mol Genet. 1996;5:1373–5. - PubMed
-
- Sambuughin N, Sivakumar K, Selenge B, et al. Autosomal dominant distal spinal muscular atrophy type V (dSMA-V) and Charcot–Marie–Tooth disease type 2D (CMT2D) segregate within a single large kindred and map to a refined region on chromosome 7p15. J Neurol Sci. 1998;161:23–8. - PubMed
-
- Silver JR. Familial spastic paraplegia with amyotrophy of the hands. Ann Hum Genet. 1966;30:69–75. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
