

Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer's disease
- PMID: 17664291
- PMCID: PMC2118680
- DOI: 10.1084/jem.20070304
Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer's disease
Abstract
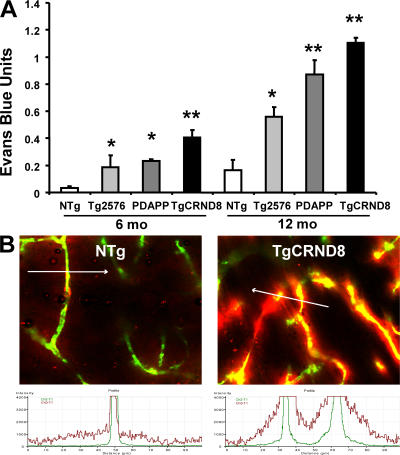
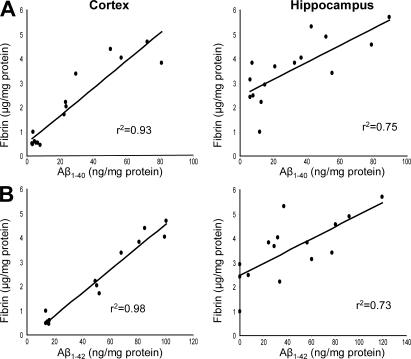
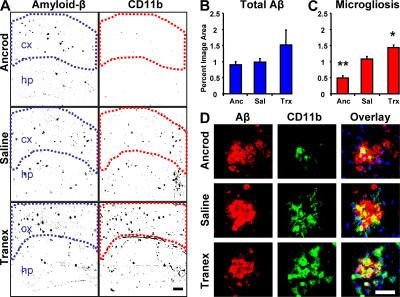
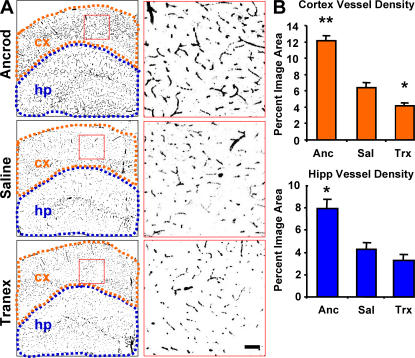
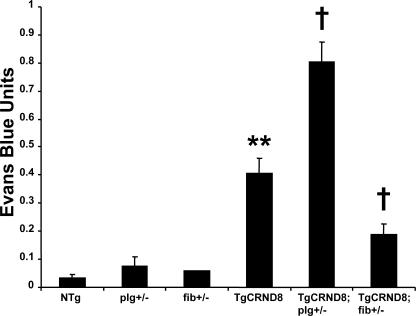
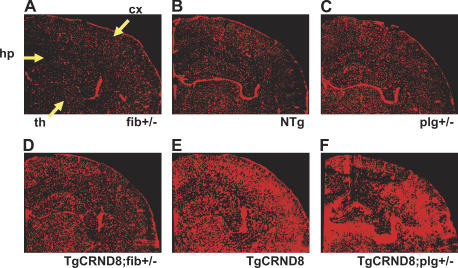
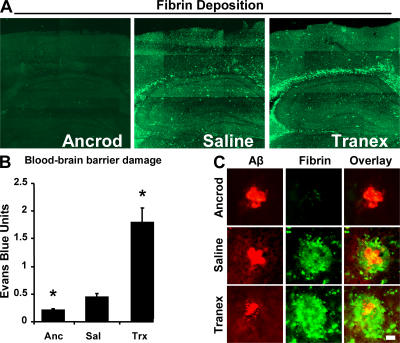
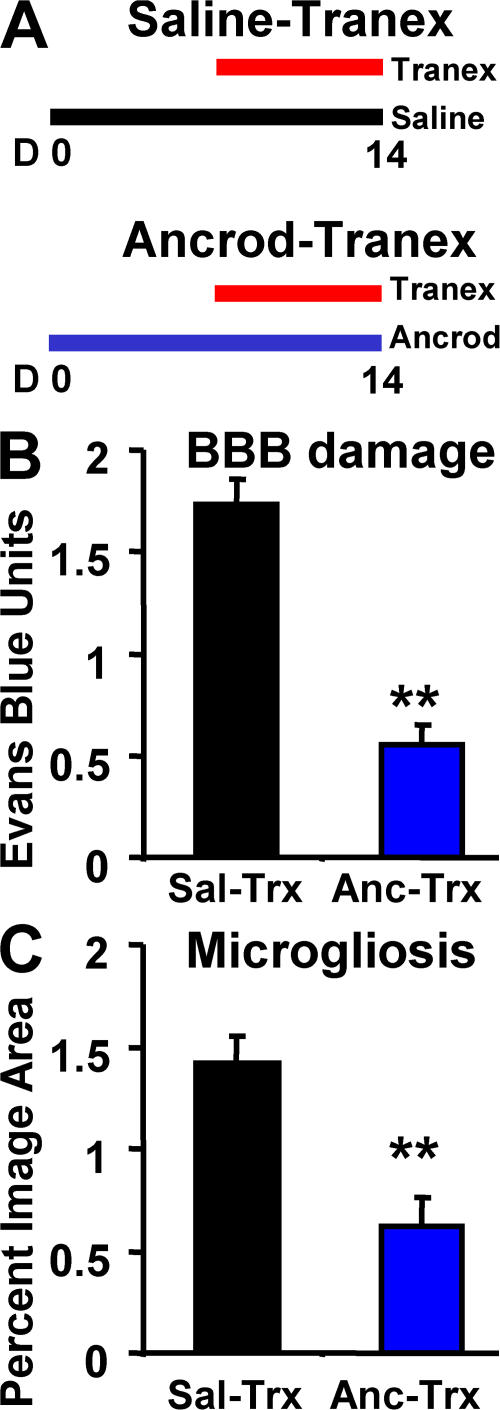
Cerebrovascular dysfunction contributes to the pathology and progression of Alzheimer's disease (AD), but the mechanisms are not completely understood. Using transgenic mouse models of AD (TgCRND8, PDAPP, and Tg2576), we evaluated blood-brain barrier damage and the role of fibrin and fibrinolysis in the progression of amyloid-beta pathology. These mouse models showed age-dependent fibrin deposition coincident with areas of blood-brain barrier permeability as demonstrated by Evans blue extravasation. Three lines of evidence suggest that fibrin contributes to the pathology. First, AD mice with only one functional plasminogen gene, and therefore with reduced fibrinolysis, have increased neurovascular damage relative to AD mice. Conversely, AD mice with only one functional fibrinogen gene have decreased blood-brain barrier damage. Second, treatment of AD mice with the plasmin inhibitor tranexamic acid aggravated pathology, whereas removal of fibrinogen from the circulation of AD mice with ancrod treatment attenuated measures of neuroinflammation and vascular pathology. Third, pretreatment with ancrod reduced the increased pathology from plasmin inhibition. These results suggest that fibrin is a mediator of inflammation and may impede the reparative process for neurovascular damage in AD. Fibrin and the mechanisms involved in its accumulation and clearance may present novel therapeutic targets in slowing the progression of AD.
Figures
References
-
- Hardy, J., and D.J. Selkoe. 2002. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 297:353–356. - PubMed
-
- de la Torre, J.C. 2004. Is Alzheimer's disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol. 3:184–190. - PubMed
-
- Farkas, E., and P.G. Luiten. 2001. Cerebral microvascular pathology in aging and Alzheimer's disease. Prog. Neurobiol. 64:575–611. - PubMed
-
- Vinters, H.V., Z.Z. Wang, and D.L. Secor. 1996. Brain parenchymal and microvascular amyloid in Alzheimer's disease. Brain Pathol. 6:179–195. - PubMed
-
- Fiala, M., Q.N. Liu, J. Sayre, V. Pop, V. Brahmandam, M.C. Graves, and H.V. Vinters. 2002. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer's disease brain and damage the blood-brain barrier. Eur. J. Clin. Invest. 32:360–371. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
