

Falling into TRAPS--receptor misfolding in the TNF receptor 1-associated periodic fever syndrome
- PMID: 17666110
- PMCID: PMC2206363
- DOI: 10.1186/ar2197
Falling into TRAPS--receptor misfolding in the TNF receptor 1-associated periodic fever syndrome
Abstract
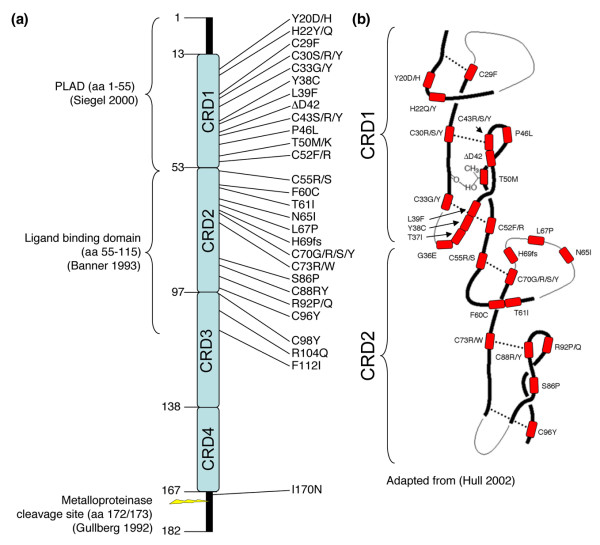
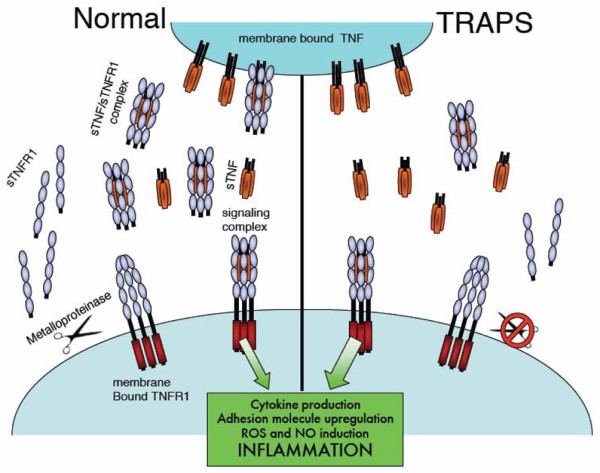
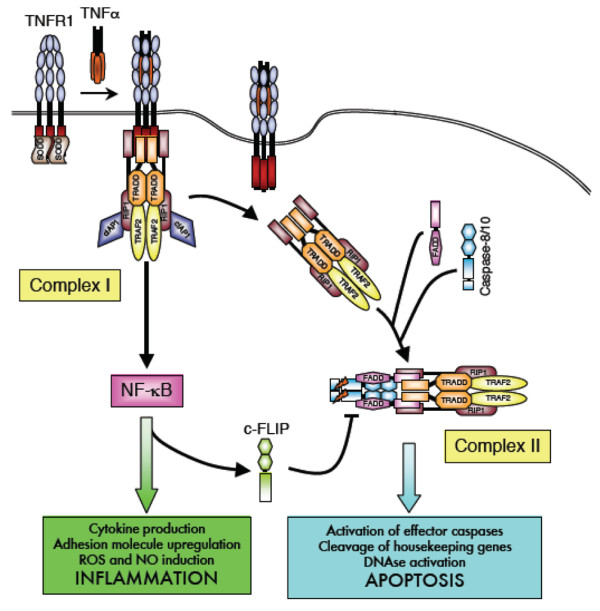
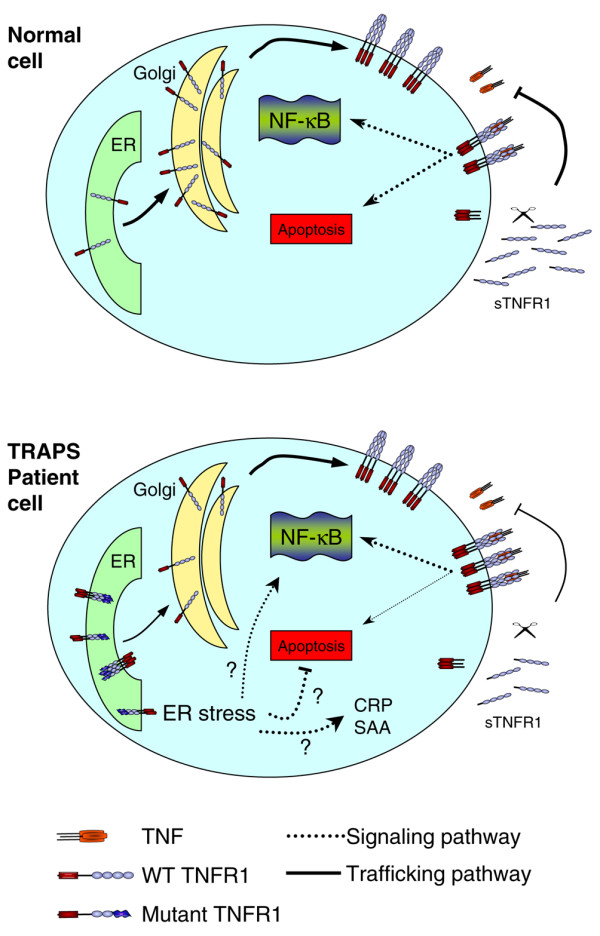
TNF receptor-associated periodic syndrome (TRAPS) is a dominantly inherited disease caused by missense mutations in the TNF receptor 1 (TNFR1) gene. Patients suffer from periodic bouts of severe abdominal pain, localised inflammation, migratory rashes, and fever. More than 40 individual mutations have been identified, all of which occur in the extracellular domain of TNFR1. In the present review we discuss new findings describing aberrant trafficking and function of TNFR1 harbouring TRAPS mutations, challenging the hypothesis that TRAPS pathology is driven by defective receptor shedding, and we suggest that TNFR1 might acquire novel functions in the endoplasmic reticulum, distinct from its role as a cell surface receptor. We also describe the clinical manifestations of TRAPS, current treatment regimens, and the widening array of patient mutations.
Figures
References
-
- Centola M, Wood G, Frucht DM, Galon J, Aringer M, Farrell C, Kingma DW, Horwitz ME, Mansfield E, Holland SM, et al. The gene for familial Mediterranean fever, MEFV, is expressed in early leukocyte development and is regulated in response to inflammatory mediators. Blood. 2000;95:3223–3231. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
