

A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN
- PMID: 17668388
- PMCID: PMC1950796
- DOI: 10.1086/519697
A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN
Abstract
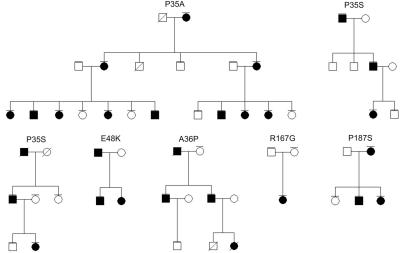
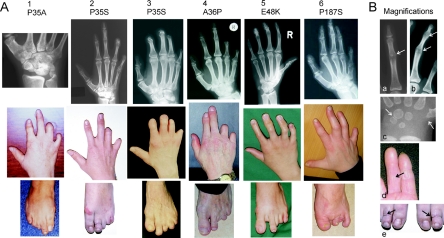
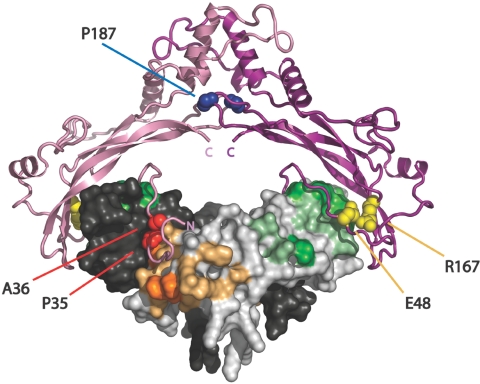
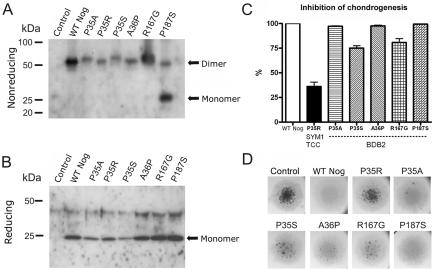
Brachydactyly type B (BDB) is characterized by terminal deficiency of fingers and toes, which is caused by heterozygous truncating mutations in the receptor tyrosine kinase-like orphan receptor 2 (ROR2) in the majority of patients. In a subset of ROR2-negative patients with BDB, clinically defined by the additional occurrence of proximal symphalangism and carpal synostosis, we identified six different point mutations (P35A, P35S, A36P, E48K, R167G, and P187S) in the bone morphogenetic protein (BMP) antagonist NOGGIN (NOG). In contrast to previously described loss-of-function mutations in NOG, which are known to cause a range of conditions associated with abnormal joint formation but without BDB, the newly identified BDB mutations do not indicate a major loss of function, as suggested by calculation of free-binding energy of the modeled NOG-GDF5 complex and functional analysis of the micromass culture system. Rather, they presumably alter NOG's ability to bind to BMPs and growth-differentiation factors (GDFs) in a subtle way, thus disturbing the intricate balance of BMP signaling. The combined features observed in this phenotypic subtype of BDB argue for a functional connection between BMP and ROR2 signaling and support previous findings of a modulating effect of ROR2 on the BMP-receptor pathway through the formation of a heteromeric complex of the receptors at the cell surface.
Figures
References
Web Resources
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for ROR2 [accession number NM_004560] and NOG [accession number NM_005450])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for BDB1, SYM1, TCC, SYNS1, and stapes ankylosis with broad thumb and toes without SYM)
References
-
- Bell J (1951) On brachydactyly and symphalangism. In: Penrose, LS (ed) Treasury of human inheritance. Vol 5. Cambridge University Press, London, United Kingdom, pp 1–31
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
