

Oncogenic activity of epidermal growth factor receptor kinase mutant alleles is enhanced by the T790M drug resistance mutation
- PMID: 17671201
- PMCID: PMC2882853
- DOI: 10.1158/0008-5472.CAN-06-4625
Oncogenic activity of epidermal growth factor receptor kinase mutant alleles is enhanced by the T790M drug resistance mutation
Abstract
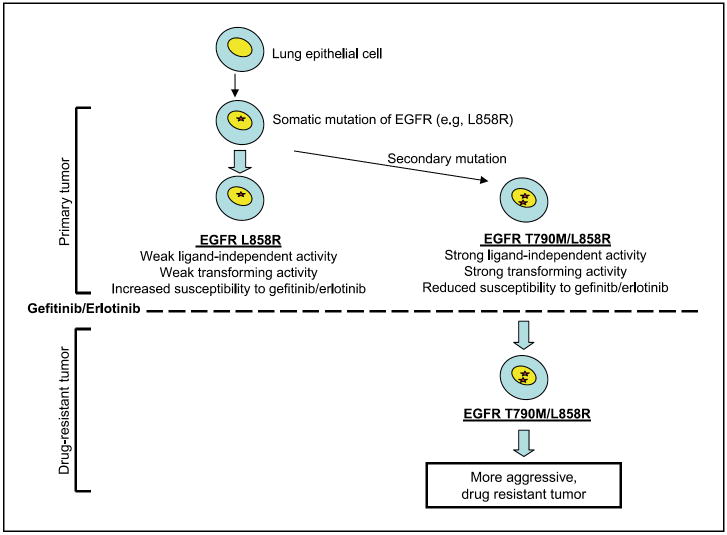
Activating mutations in the epidermal growth factor receptor (EGFR) characterize a subset of non-small cell lung cancers (NSCLC) with extraordinary sensitivity to targeted tyrosine kinase inhibitors (TKI). A single secondary EGFR mutation, T790M, arising in cis with the primary activating mutation, confers acquired resistance to these drugs. However, the T790M mutation is also detected in the absence of drug selection, suggesting that it may provide a growth advantage. We show here that although T790M alone has only a modest effect on EGFR function, when combined with the characteristic activating mutations L858R or del746-750, it results in a dramatic enhancement of EGFR activity. The double mutants show potent ligand-independent receptor autophosphorylation associated with altered cellular phenotypes, soft agar colony formation, and tumorigenesis in nude mice. The significant gain-of-function properties of these double mutants may explain their initial presence before drug selection and their rapid selection as the single drug resistance mutation during therapy with gefitinib/erlotinib, and suggests that they may contribute to the adverse clinical course of TKI-resistant NSCLC.
Figures

References
-
- Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. see comment. - PubMed
-
- Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. see comment. - PubMed
-
- Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate antiapoptotic pathways. Science. 2004;305:1163–7. - PubMed
-
- Weinstein IB. Cancer. Addiction to oncogenes—the Achilles heal of cancer. Science. 2002;297:63–4. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
