

Evolution of naturally occurring 5'non-coding region variants of Hepatitis C virus in human populations of the South American region
- PMID: 17683527
- PMCID: PMC1973069
- DOI: 10.1186/1743-422X-4-79
Evolution of naturally occurring 5'non-coding region variants of Hepatitis C virus in human populations of the South American region
Abstract
Background: Hepatitis C virus (HCV) has been the subject of intense research and clinical investigation as its major role in human disease has emerged. Previous and recent studies have suggested a diversification of type 1 HCV in the South American region. The degree of genetic variation among HCV strains circulating in Bolivia and Colombia is currently unknown. In order to get insight into these matters, we performed a phylogenetic analysis of HCV 5' non-coding region (5'NCR) sequences from strains isolated in Bolivia, Colombia and Uruguay, as well as available comparable sequences of HCV strains isolated in South America.
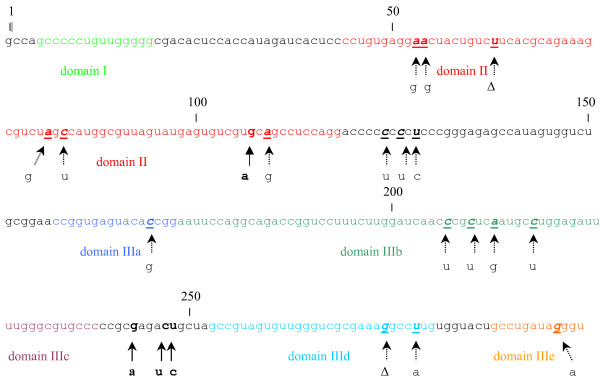
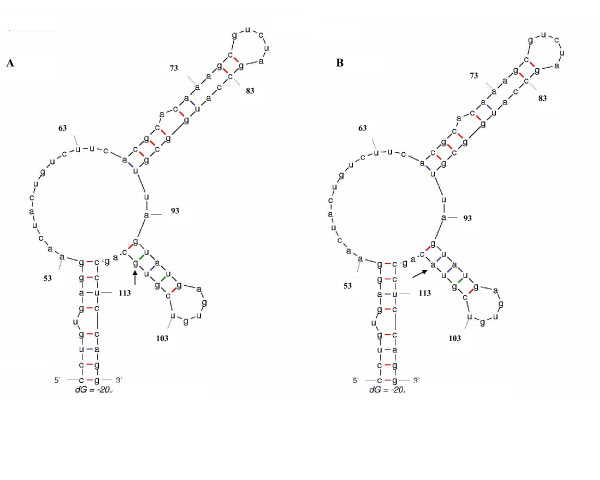
Methods: Phylogenetic tree analysis was performed using the neighbor-joining method under a matrix of genetic distances established under the Kimura-two parameter model. Signature pattern analysis, which identifies particular sites in nucleic acid alignments of variable sequences that are distinctly representative relative to a background set, was performed using the method of Korber & Myers, as implemented in the VESPA program. Prediction of RNA secondary structures was done by the method of Zuker & Turner, as implemented in the mfold program.
Results: Phylogenetic tree analysis of HCV strains isolated in the South American region revealed the presence of a distinct genetic lineage inside genotype 1. Signature pattern analysis revealed that the presence of this lineage is consistent with the presence of a sequence signature in the 5'NCR of HCV strains isolated in South America. Comparisons of these results with the ones found for Europe or North America revealed that this sequence signature is characteristic of the South American region.
Conclusion: Phylogentic analysis revealed the presence of a sequence signature in the 5'NCR of type 1 HCV strains isolated in South America. This signature is frequent enough in type 1 HCV populations circulating South America to be detected in a phylogenetic tree analysis as a distinct type 1 sub-population. The coexistence of distinct type 1 HCV subpopulations is consistent with quasispecies dynamics, and suggests that multiple coexisting subpopulations may allow the virus to adapt to its human host populations.
Figures


Similar articles
-
Diversification of hepatitis C viruses in South America reveals a novel genetic lineage.Arch Virol. 2001 Aug;146(8):1623-9. doi: 10.1007/s007050170084. Arch Virol. 2001. PMID: 11676423
-
Analysis of genetic heterogeneity of hepatitis C viruses in Central America reveals a novel genetic lineage. Brief report.Arch Virol. 2002 Nov;147(11):2239-46. doi: 10.1007/s00705-002-0869-4. Arch Virol. 2002. PMID: 12417958
-
Analysis of genetic variability of Indian isolates of Hepatitis C virus.Arch Virol. 2004 Jun;149(6):1185-92. doi: 10.1007/s00705-003-0286-3. Epub 2004 Mar 1. Arch Virol. 2004. PMID: 15168204
-
Evidence of increasing diversification of hepatitis C viruses.J Gen Virol. 1999 Jun;80 ( Pt 6):1377-1382. doi: 10.1099/0022-1317-80-6-1377. J Gen Virol. 1999. PMID: 10374954
-
Genetic diversity and evolution of hepatitis C virus in the Latin American region.J Clin Virol. 2005 Dec;34 Suppl 2:S1-7. doi: 10.1016/s1386-6532(05)00393-8. J Clin Virol. 2005. PMID: 16461234 Review.
Cited by
-
Virological surveillance, molecular phylogeny, and evolutionary dynamics of hepatitis C virus subtypes 1a and 4a isolates in patients from Saudi Arabia.Saudi J Biol Sci. 2021 Mar;28(3):1664-1677. doi: 10.1016/j.sjbs.2020.11.089. Epub 2020 Dec 8. Saudi J Biol Sci. 2021. PMID: 33732052 Free PMC article.
-
Genetic history of hepatitis C virus in Venezuela: high diversity and long time of evolution of HCV genotype 2.PLoS One. 2010 Dec 13;5(12):e14315. doi: 10.1371/journal.pone.0014315. PLoS One. 2010. PMID: 21179440 Free PMC article.
References
-
- Simmonds P, Bukh J, Combet C, Deleage G, Enomoto N, Feinstone S, Halfon P, Inchauspe G, Kuiken C, Maertens G, Mizokami M, Murphy DG, Okamoto H, Pawlotsky JM, Penin F, Sablon E, Shin-IT , Stuyver LJ, Thiel HJ, Viazov S, Weiner AD, Widell A. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hematology. 2005;42:962–973. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
