

A novel DSPP mutation is associated with type II dentinogenesis imperfecta in a Chinese family
- PMID: 17686168
- PMCID: PMC1995191
- DOI: 10.1186/1471-2350-8-52
A novel DSPP mutation is associated with type II dentinogenesis imperfecta in a Chinese family
Abstract
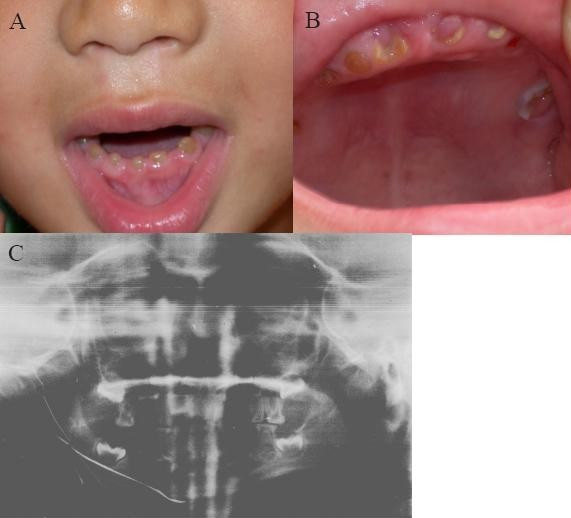
Background: Hereditary defects of tooth dentin are classified into two main groups: dentin dysplasia (DD) (types I and II) and dentinogenesis imperfecta (DGI) (types I, II, and III). Type II DGI is one of the most common tooth defects with an autosomal dominant mode of inheritance. One disease-causing gene, the dentin sialophosphoprotein (DSPP) gene, has been reported for type II DGI.
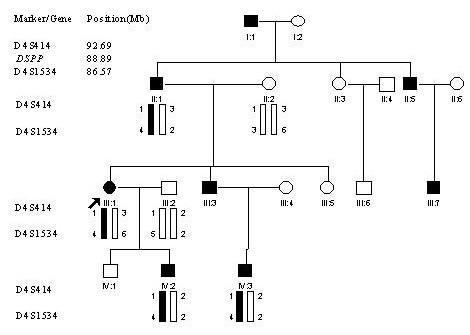
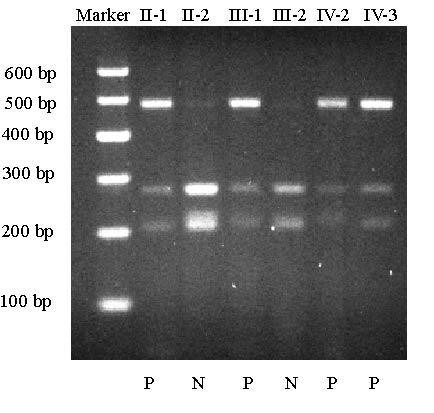
Methods: In this study, we characterized a four-generation Chinese family with type II DGI that consists of 18 living family members, including 8 affected individuals. Linkage analysis with polymorphic markers D4S1534 and D4S414 that span the DSPP gene showed that the family is linked to DSPP. All five exons and exon-intron boundaries of DSPP were sequenced in members of type II DGI family.
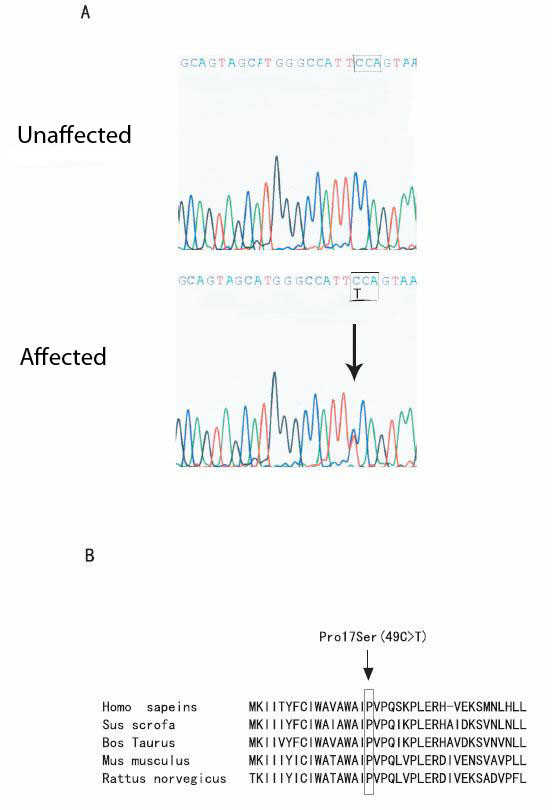
Results: Direct DNA sequence analysis identified a novel mutation (c.49C-->T, p.Pro17Ser) in exon 1 of the DSPP gene. The mutation spot, the Pro17 residue, is the second amino acid of the mature DSP protein, and highly conserved during evolution. The mutation was identified in all affected individuals, but not in normal family members and 100 controls.
Conclusion: These results suggest that mutation p.Pro17Ser causes type II DGI in the Chinese family. This study identifies a novel mutation in the DSPP gene, and expands the spectrum of mutations that cause DGI.
Figures
References
-
- Witkop CJ., Jr Hereditary defects in enamel and dentin. Acta Genet Stat Med. 1957;7:236–239. - PubMed
-
- MacDougall M, Simmons D, Luan X, Nydegger J, Feng J, Gu TT. Dentin phosphoprotein and dentin sialoprotein are cleavage products expressed from a single transcript coded by a gene on human chromosome 4. Dentin phosphoprotein DNA sequence determination. J Biol Chem. 1997;272:835–842. doi: 10.1074/jbc.272.2.835. - DOI - PubMed
-
- Ritchie HH, Hou H, Veis A, Butler WT. Cloning and sequence determination of rat dentin sialoprotein, a novel dentin protein. J Biol Chem. 1994;269:3698–3702. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
