

Toward a universal microarray: prediction of gene expression through nearest-neighbor probe sequence identification
- PMID: 17686789
- PMCID: PMC1976448
- DOI: 10.1093/nar/gkm549
Toward a universal microarray: prediction of gene expression through nearest-neighbor probe sequence identification
Abstract
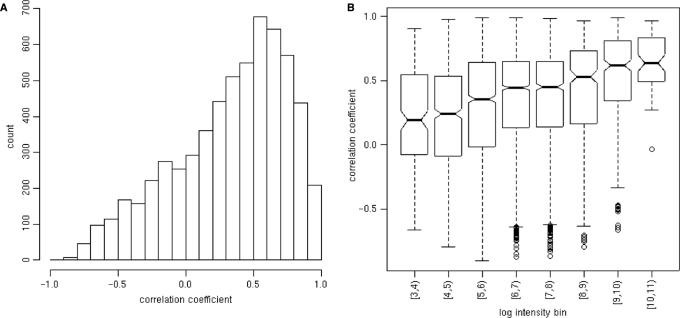
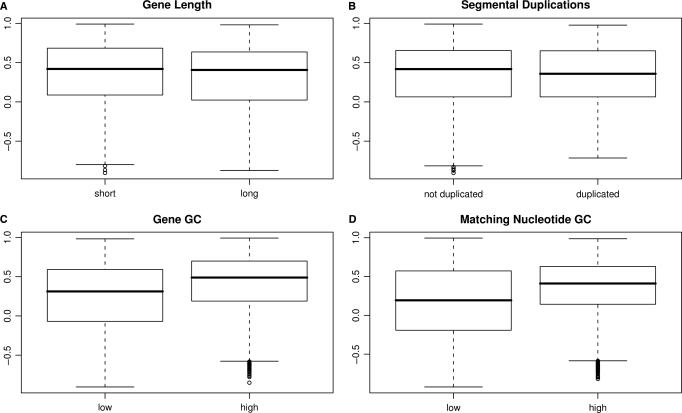
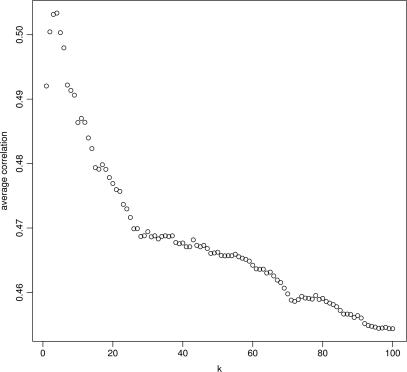
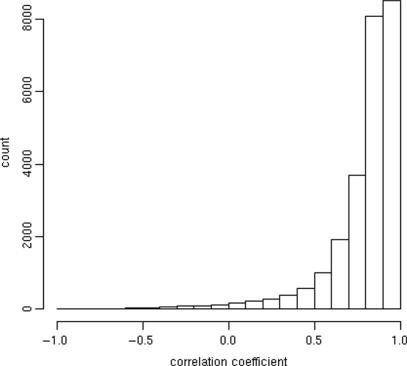
A generic DNA microarray design applicable to any species would greatly benefit comparative genomics. We have addressed the feasibility of such a design by leveraging the great feature densities and relatively unbiased nature of genomic tiling microarrays. Specifically, we first divided each Homo sapiens Refseq-derived gene's spliced nucleotide sequence into all of its possible contiguous 25 nt subsequences. For each of these 25 nt subsequences, we searched a recent human transcript mapping experiment's probe design for the 25 nt probe sequence having the fewest mismatches with the subsequence, but that did not match the subsequence exactly. Signal intensities measured with each gene's nearest-neighbor features were subsequently averaged to predict their gene expression levels in each of the experiment's thirty-three hybridizations. We examined the fidelity of this approach in terms of both sensitivity and specificity for detecting actively transcribed genes, for transcriptional consistency between exons of the same gene, and for reproducibility between tiling array designs. Taken together, our results provide proof-of-principle for probing nucleic acid targets with off-target, nearest-neighbor features.
Figures
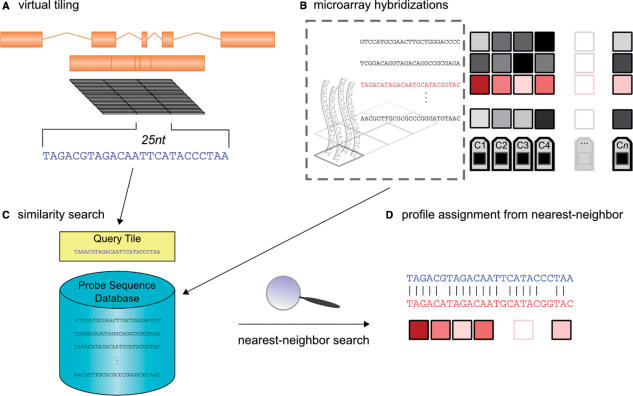
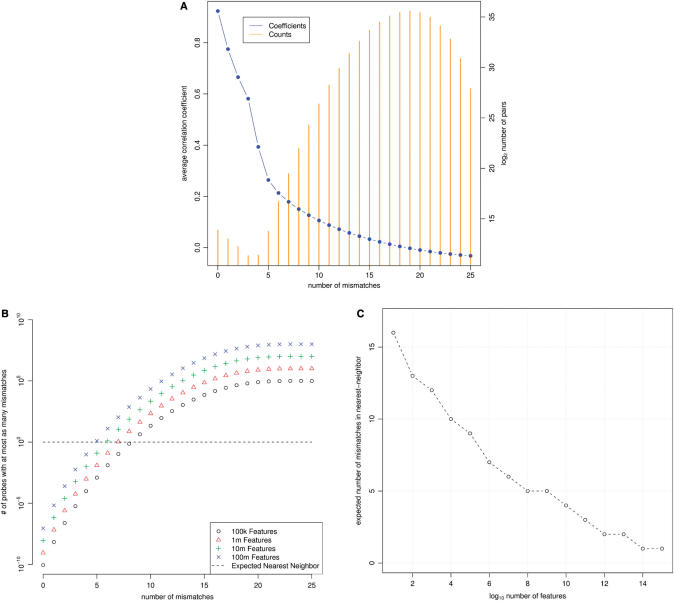
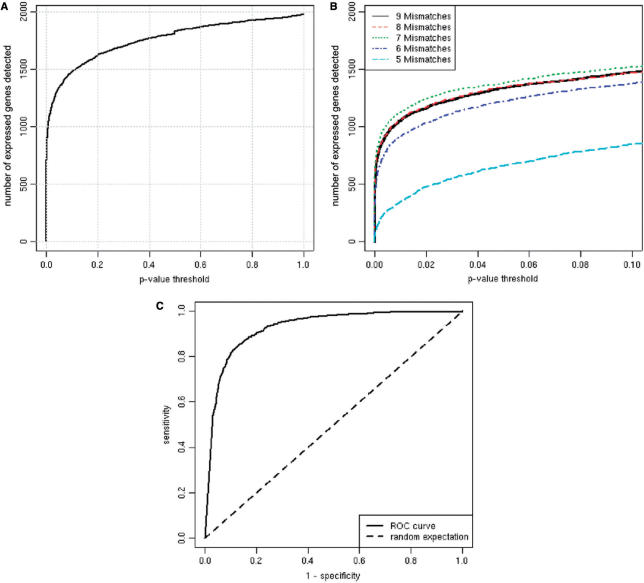
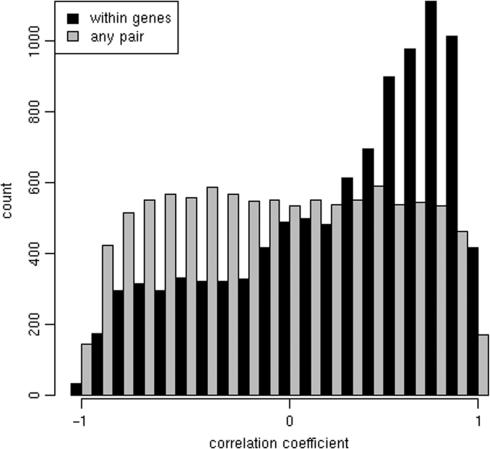
References
-
- Selinger DW, Cheung KJ, Mei R, Johansson EM, Richmond CS, Blattner FR, Lockhart DJ, Church GM. RNA expression analysis using a 30 base pair resolution Escherichia coli genome array. Nat. Biotechnol. 2000;18:1262–1268. - PubMed
-
- Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, Zhu X, Rinn JL, Tongprasit W, Samanta M, et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–2246. - PubMed
-
- Kapranov P, Cawley SE, Drenkow J, Bekiranov S, Strausberg RL, Fodor SPA, Gingeras TR. Large-scale transcriptional activity in chromosomes 21 and 22. Science. 2002;296:916–919. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
