

Structural footprinting in protein structure comparison: the impact of structural fragments
- PMID: 17688700
- PMCID: PMC2082327
- DOI: 10.1186/1472-6807-7-53
Structural footprinting in protein structure comparison: the impact of structural fragments
Abstract
Background: One approach for speeding-up protein structure comparison is the projection approach, where a protein structure is mapped to a high-dimensional vector and structural similarity is approximated by distance between the corresponding vectors. Structural footprinting methods are projection methods that employ the same general technique to produce the mapping: first select a representative set of structural fragments as models and then map a protein structure to a vector in which each dimension corresponds to a particular model and "counts" the number of times the model appears in the structure. The main difference between any two structural footprinting methods is in the set of models they use; in fact a large number of methods can be generated by varying the type of structural fragments used and the amount of detail in their representation. How do these choices affect the ability of the method to detect various types of structural similarity?
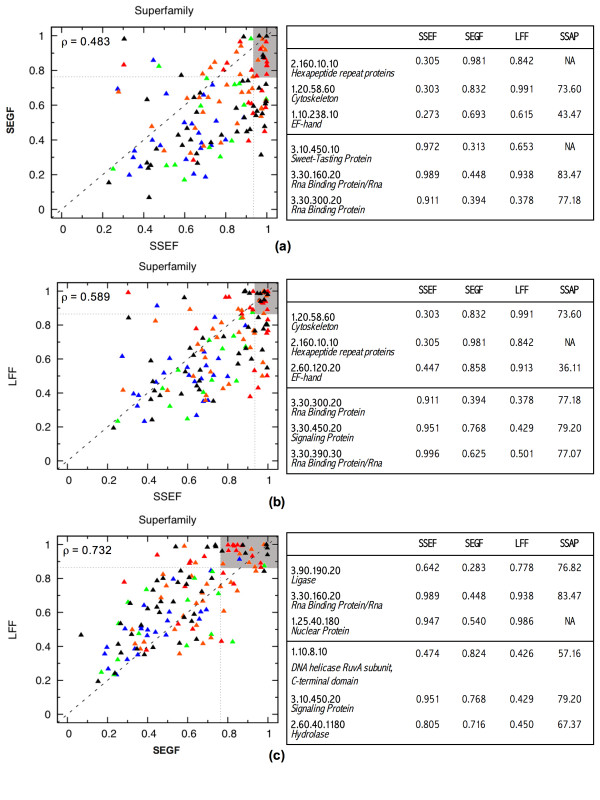
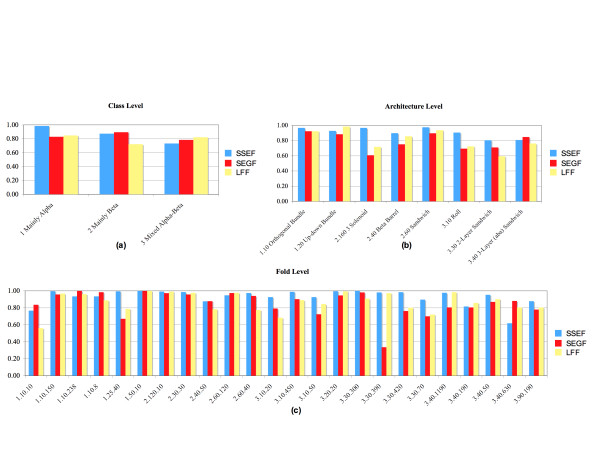
Results: To answer this question we benchmarked three structural footprinting methods that vary significantly in their selection of models against the CATH database. In the first set of experiments we compared the methods' ability to detect structural similarity characteristic of evolutionarily related structures, i.e., structures within the same CATH superfamily. In the second set of experiments we tested the methods' agreement with the boundaries imposed by classification groups at the Class, Architecture, and Fold levels of the CATH hierarchy.
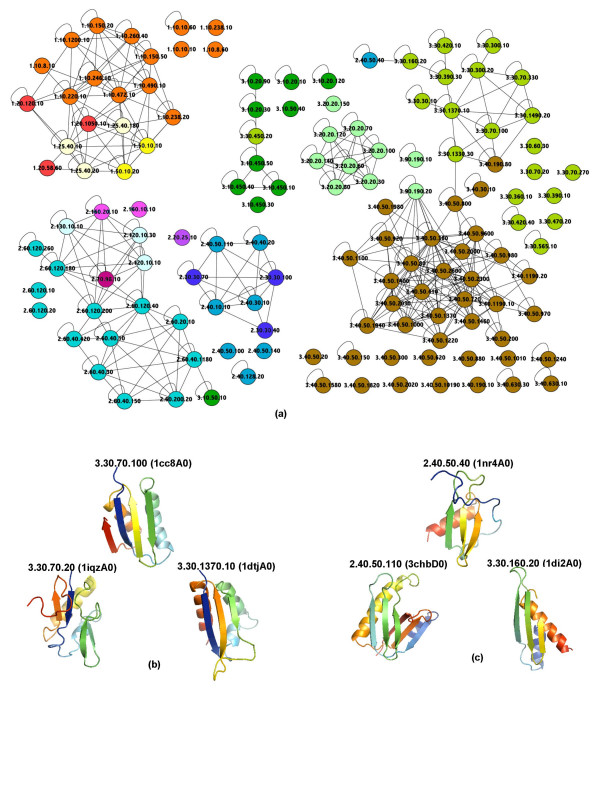
Conclusion: In both experiments we found that the method which uses secondary structure information has the best performance on average, but no one method performs consistently the best across all groups at a given classification level. We also found that combining the methods' outputs significantly improves the performance. Moreover, our new techniques to measure and visualize the methods' agreement with the CATH hierarchy, including the threshholded affinity graph, are useful beyond this work. In particular, they can be used to expose a similar composition of different classification groups in terms of structural fragments used by the method and thus provide an alternative demonstration of the continuous nature of the protein structure universe.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
