

ORF73-null murine gammaherpesvirus 68 reveals roles for mLANA and p53 in virus replication
- PMID: 17699571
- PMCID: PMC2168792
- DOI: 10.1128/JVI.00111-07
ORF73-null murine gammaherpesvirus 68 reveals roles for mLANA and p53 in virus replication
Abstract
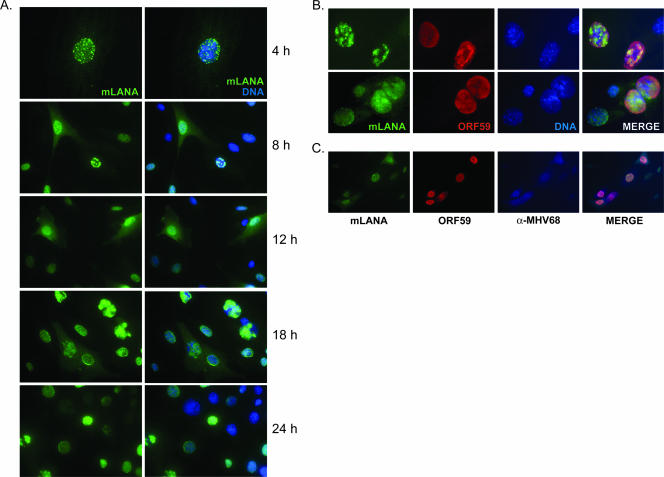
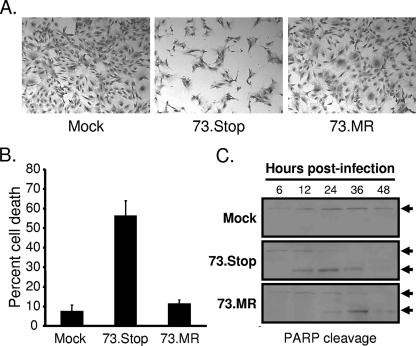
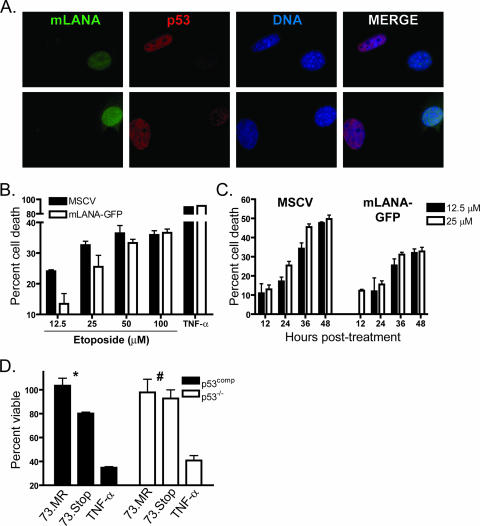
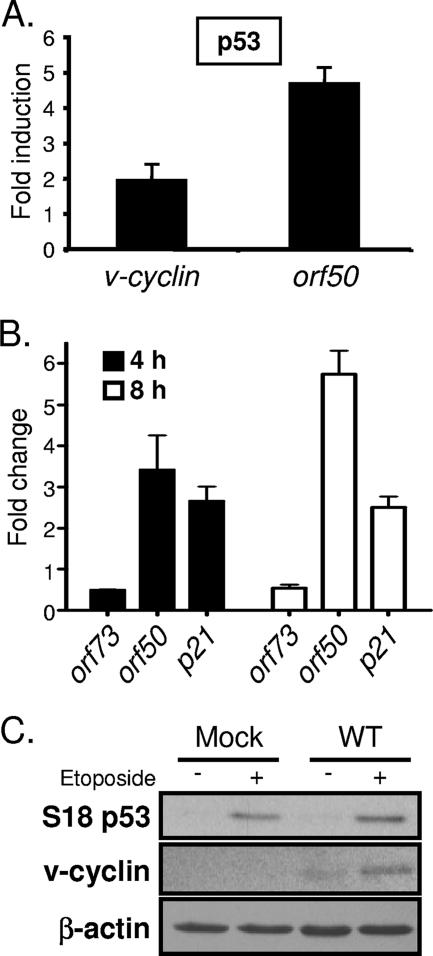
Gammaherpesviruses establish lifelong, latent infections in host lymphocytes, during which a limited subset of viral gene products facilitates maintenance of the viral episome. Among the gamma-2-herpesvirus (rhadinovirus) subfamily, this includes expression of the conserved ORF73-encoded LANA proteins. We previously demonstrated by loss-of-function mutagenesis that the murine gammaherpesvirus 68 (MHV68) ORF73 gene product, mLANA, is required for the establishment of latency following intranasal inoculation of mice (N. J. Moorman, D. O. Willer, and S. H. Speck, J. Virol. 77:10295-10303, 2003). mLANA-deficient viruses also exhibited a defect in acute virus replication in the lungs of infected mice. The latter observation led us to examine the role of mLANA in productive viral replication. We assessed the capacity of mLANA-deficient virus (73.Stop) to replicate in cell culture at low multiplicities of infection (MOIs) and found that 73.Stop growth was impaired in murine fibroblasts but not in Vero cells. A recombinant virus expressing an mLANA-green fluorescent protein (GFP) fusion revealed that mLANA is expressed throughout the virus replication cycle. In addition, 73.Stop infection of murine fibroblasts at high MOIs was substantially more cytotoxic than infection with a genetically repaired marker rescue virus (73.MR), a phenotype that correlated with enhanced kinetics of viral gene expression and increased activation of p53. Notably, augmented cell death, viral gene expression, and p53 induction were independent of viral DNA replication. Expression of a mLANA-GFP fusion protein in fibroblasts correlated with both reduced p53 stabilization and reduced cell death following treatment with p53-inducing agonists. In agreement, accentuated cell death associated with 73.Stop infection was reduced in p53-deficient murine embryonic fibroblasts. Additionally, replication of 73.Stop in p53-deficient cells was restored to levels comparable to those of 73.MR. More remarkably, the absence of p53 led to an overall delay in replication for both 73.Stop and 73.MR viruses, which correlated with delayed viral gene expression, indicating a role for p53 in MHV68 replication. Consistent with these findings, the expression of replication-promoting viral genes was positively influenced by p53 overexpression or treatment with the p53 agonist etoposide. Overall, these data demonstrate the importance of mLANA in MHV68 replication and suggest that LANA proteins limit the induction of cellular stress responses to regulate the viral gene expression cascade and limit host cell injury.
Figures





Similar articles
-
Murine Gammaherpesvirus 68 LANA and SOX Homologs Counteract ATM-Driven p53 Activity during Lytic Viral Replication.J Virol. 2015 Dec 16;90(5):2571-85. doi: 10.1128/JVI.02867-15. J Virol. 2015. PMID: 26676792 Free PMC article.
-
Identification of Viral and Host Proteins That Interact with Murine Gammaherpesvirus 68 Latency-Associated Nuclear Antigen during Lytic Replication: a Role for Hsc70 in Viral Replication.J Virol. 2015 Nov 18;90(3):1397-413. doi: 10.1128/JVI.02022-15. Print 2016 Feb 1. J Virol. 2015. PMID: 26581985 Free PMC article.
-
Murine Gammaherpesvirus 68 Expressing Kaposi Sarcoma-Associated Herpesvirus Latency-Associated Nuclear Antigen (LANA) Reveals both Functional Conservation and Divergence in LANA Homologs.J Virol. 2017 Sep 12;91(19):e00992-17. doi: 10.1128/JVI.00992-17. Print 2017 Oct 1. J Virol. 2017. PMID: 28747501 Free PMC article.
-
Gamma herpesviruses: pathogenesis of infection and cell signaling.Folia Microbiol (Praha). 2003;48(3):291-318. doi: 10.1007/BF02931360. Folia Microbiol (Praha). 2003. PMID: 12879740 Review.
-
Modulation of the ARF-p53 pathway by the small DNA tumor viruses.Cell Cycle. 2005 Mar;4(3):449-52. doi: 10.4161/cc.4.3.1555. Epub 2005 Mar 24. Cell Cycle. 2005. PMID: 15738654 Review.
Cited by
-
An essential role for γ-herpesvirus latency-associated nuclear antigen homolog in an acute lymphoproliferative disease of cattle.Proc Natl Acad Sci U S A. 2013 May 21;110(21):E1933-42. doi: 10.1073/pnas.1216531110. Epub 2013 Apr 29. Proc Natl Acad Sci U S A. 2013. PMID: 23630278 Free PMC article.
-
Phosphorylation of the chromatin binding domain of KSHV LANA.PLoS Pathog. 2012;8(10):e1002972. doi: 10.1371/journal.ppat.1002972. Epub 2012 Oct 18. PLoS Pathog. 2012. PMID: 23093938 Free PMC article.
-
Murine Gammaherpesvirus 68 LANA and SOX Homologs Counteract ATM-Driven p53 Activity during Lytic Viral Replication.J Virol. 2015 Dec 16;90(5):2571-85. doi: 10.1128/JVI.02867-15. J Virol. 2015. PMID: 26676792 Free PMC article.
-
Identification of Viral and Host Proteins That Interact with Murine Gammaherpesvirus 68 Latency-Associated Nuclear Antigen during Lytic Replication: a Role for Hsc70 in Viral Replication.J Virol. 2015 Nov 18;90(3):1397-413. doi: 10.1128/JVI.02022-15. Print 2016 Feb 1. J Virol. 2015. PMID: 26581985 Free PMC article.
-
Unbiased mutagenesis of MHV68 LANA reveals a DNA-binding domain required for LANA function in vitro and in vivo.PLoS Pathog. 2012 Sep;8(9):e1002906. doi: 10.1371/journal.ppat.1002906. Epub 2012 Sep 6. PLoS Pathog. 2012. PMID: 22969427 Free PMC article.
References
-
- An, F. Q., N. Compitello, E. Horwitz, M. Sramkoski, E. S. Knudsen, and R. Renne. 2005. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus modulates cellular gene expression and protects lymphoid cells from p16 INK4A-induced cell cycle arrest. J. Biol. Chem. 280:3862-3874. - PubMed
-
- Baptiste, N., and C. Prives. 2004. p53 in the cytoplasm: a question of overkill? Cell 116:487-489. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
