

Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study
- PMID: 17701892
- PMCID: PMC1950837
- DOI: 10.1086/520125
Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study
Abstract
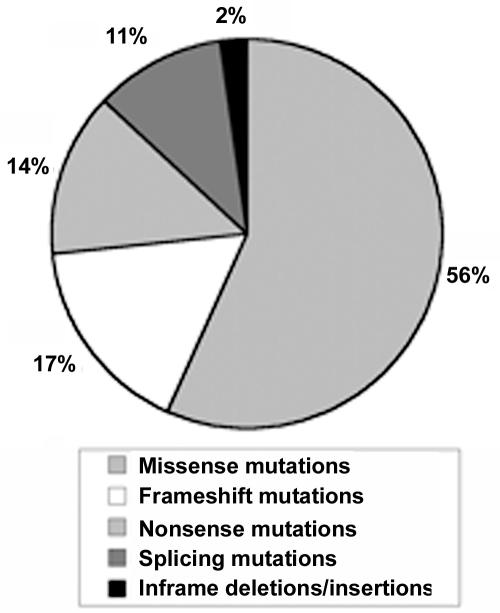
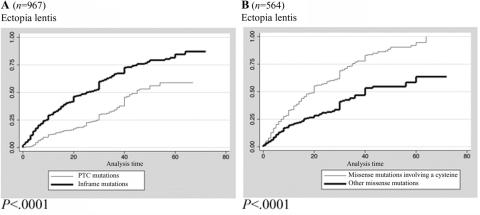
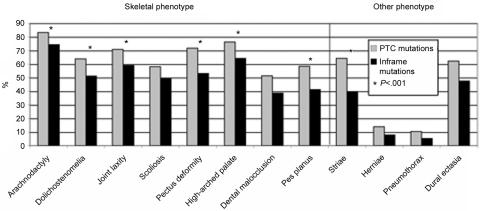
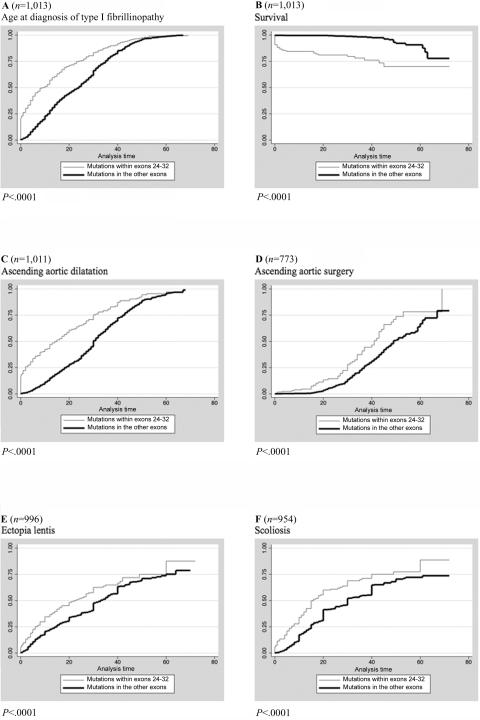
Mutations in the fibrillin-1 (FBN1) gene cause Marfan syndrome (MFS) and have been associated with a wide range of overlapping phenotypes. Clinical care is complicated by variable age at onset and the wide range of severity of aortic features. The factors that modulate phenotypical severity, both among and within families, remain to be determined. The availability of international FBN1 mutation Universal Mutation Database (UMD-FBN1) has allowed us to perform the largest collaborative study ever reported, to investigate the correlation between the FBN1 genotype and the nature and severity of the clinical phenotype. A range of qualitative and quantitative clinical parameters (skeletal, cardiovascular, ophthalmologic, skin, pulmonary, and dural) was compared for different classes of mutation (types and locations) in 1,013 probands with a pathogenic FBN1 mutation. A higher probability of ectopia lentis was found for patients with a missense mutation substituting or producing a cysteine, when compared with other missense mutations. Patients with an FBN1 premature termination codon had a more severe skeletal and skin phenotype than did patients with an inframe mutation. Mutations in exons 24-32 were associated with a more severe and complete phenotype, including younger age at diagnosis of type I fibrillinopathy and higher probability of developing ectopia lentis, ascending aortic dilatation, aortic surgery, mitral valve abnormalities, scoliosis, and shorter survival; the majority of these results were replicated even when cases of neonatal MFS were excluded. These correlations, found between different mutation types and clinical manifestations, might be explained by different underlying genetic mechanisms (dominant negative versus haploinsufficiency) and by consideration of the two main physiological functions of fibrillin-1 (structural versus mediator of TGF beta signalling). Exon 24-32 mutations define a high-risk group for cardiac manifestations associated with severe prognosis at all ages.
Figures
References
Web Resources
-
- Kristine Yu's Web site, http://cmgm.stanford.edu/biochem218/Projects%202001/Yu.pdf (for paper entitled “Theoretical Determination of Amino Acid Substitution Groups Based on Qulaitiative Physicochemical Properties”)
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for MFS, FBN1, TGFBR2, isolated ectopia lentis, and Weill-Marchesani syndrome)
-
- UMD, http://www.umd.be/
References
-
- Pyeritz RE (1993) Marfan syndrome: current and future clinical and genetic management of cardiovascular manifestations. Semin Thorac Cardiovasc Surg 5:11–16 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
