

DLX5 and DLX6 expression is biallelic and not modulated by MeCP2 deficiency
- PMID: 17701895
- PMCID: PMC1950824
- DOI: 10.1086/520063
DLX5 and DLX6 expression is biallelic and not modulated by MeCP2 deficiency
Abstract
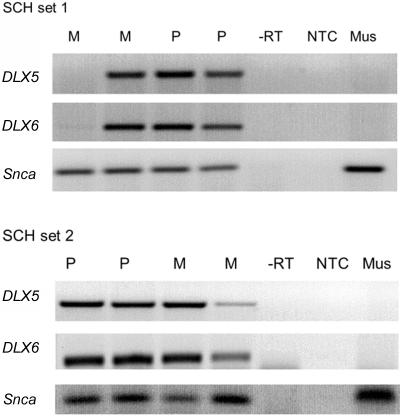
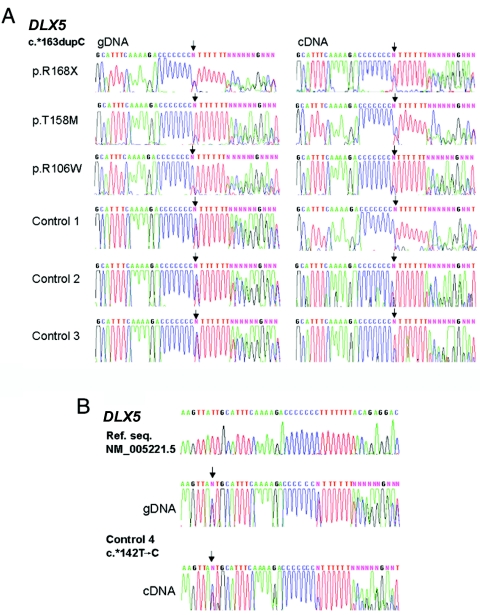
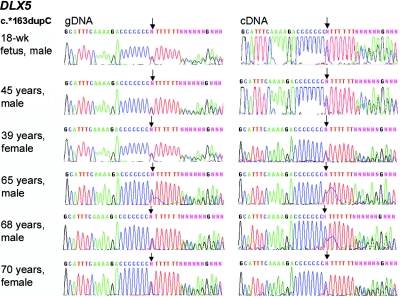
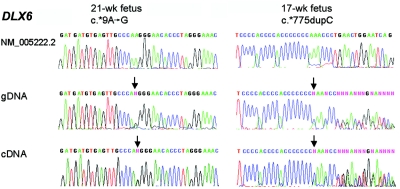
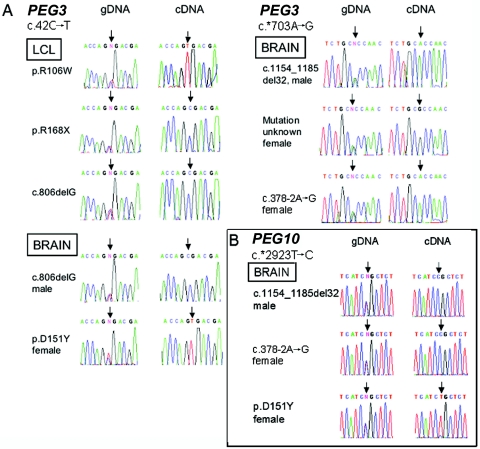
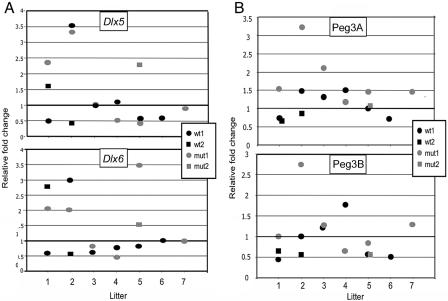
Mutations in MECP2 and Mecp2 (encoding methyl-CpG binding protein 2 [MeCP2]) cause distinct neurological phenotypes in humans and mice, respectively, but the molecular pathology is unclear. Recent literature claimed that the developmental homeobox gene DLX5 is imprinted and that its imprinting status is modulated by MeCP2, leading to biallelic expression in Rett syndrome and twofold overexpression of Dlx5 and Dlx6 in Mecp2-null mice. The conclusion that DLX5 is a direct target of MeCP2 has implications for research on the molecular bases of Rett syndrome, autism, and genomic imprinting. Attempting to replicate the reported data, we evaluated allele-specific expression of DLX5 and DLX6 in mouse x human somatic cell hybrids, lymphoblastoid cell lines, and frontal cortex from controls and individuals with MECP2 mutations. We identified novel single-nucleotide polymorphisms in DLX5 and DLX6, enabling the first imprinting studies of DLX6. We found that DLX5 and DLX6 are biallelically expressed in somatic cell hybrids and in human cell lines and brain, with no differences between affected and control samples. We also determined expression levels of Dlx5 and Dlx6 in forebrain from seven male Mecp2-mutant mice and eight wild-type littermates by real-time quantitative reverse-transcriptase polymerase chain reaction assays. Expression of Dlx5 and Dlx6, as well as of the imprinted gene Peg3, in mouse forebrain was highly variable, with no consistent differences between Mecp2-null mutants and controls. We conclude that DLX5 and DLX6 are not imprinted in humans and are not likely to be direct targets of MeCP2 modulation. In contrast, the imprinting status of PEG3 and PEG10 is maintained in MeCP2-deficient tissues. Our results confirm that MeCP2 plays no role in the maintenance of genomic imprinting and add PEG3 and PEG10 to the list of studied imprinted genes.
Figures
Similar articles
-
Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome.Nat Genet. 2005 Jan;37(1):31-40. doi: 10.1038/ng1491. Epub 2004 Dec 19. Nat Genet. 2005. PMID: 15608638
-
Imprinting status of paternally imprinted DLX5 gene in Japanese patients with Rett syndrome.Brain Dev. 2007 Sep;29(8):491-5. doi: 10.1016/j.braindev.2007.01.006. Epub 2007 Mar 23. Brain Dev. 2007. PMID: 17363207
-
Expression analysis and mutation detection of DLX5 and DLX6 in autism.Brain Dev. 2010 Feb;32(2):98-104. doi: 10.1016/j.braindev.2008.12.021. Epub 2009 Feb 4. Brain Dev. 2010. PMID: 19195802
-
Association by guilt: identification of DLX5 as a target for MeCP2 provides a molecular link between genomic imprinting and Rett syndrome.Bioessays. 2005 Jul;27(7):676-80. doi: 10.1002/bies.20266. Bioessays. 2005. PMID: 15954098 Review.
-
The Odyssey of MeCP2 and parental imprinting.Epigenetics. 2007 Jan-Mar;2(1):5-10. doi: 10.4161/epi.2.1.3697. Epub 2006 Dec 12. Epigenetics. 2007. PMID: 17965611 Free PMC article. Review.
Cited by
-
Balanced gene regulation by an embryonic brain ncRNA is critical for adult hippocampal GABA circuitry.Nat Neurosci. 2009 Aug;12(8):1020-7. doi: 10.1038/nn.2371. Epub 2009 Jul 20. Nat Neurosci. 2009. PMID: 19620975 Free PMC article.
-
The MBD protein family-reading an epigenetic mark?Mutat Res. 2008 Dec 1;647(1-2):39-43. doi: 10.1016/j.mrfmmm.2008.07.007. Epub 2008 Jul 22. Mutat Res. 2008. PMID: 18692077 Free PMC article. Review.
-
Differentially methylated regions in maternal and paternal uniparental disomy for chromosome 7.Epigenetics. 2014 Mar;9(3):351-65. doi: 10.4161/epi.27160. Epub 2013 Nov 18. Epigenetics. 2014. PMID: 24247273 Free PMC article.
-
SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice.PLoS One. 2008 Mar 5;3(3):e1709. doi: 10.1371/journal.pone.0001709. PLoS One. 2008. PMID: 18320030 Free PMC article.
-
Analysis of neonatal brain lacking ATRX or MeCP2 reveals changes in nucleosome density, CTCF binding and chromatin looping.Nucleic Acids Res. 2014 Jul;42(13):8356-68. doi: 10.1093/nar/gku564. Epub 2014 Jul 2. Nucleic Acids Res. 2014. PMID: 24990380 Free PMC article.
References
Web Resources
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for DLX5 [accession number NM_005221.5], DLX6 [accession number NM_005222.2], and Dlx6 [accession number BC114342.1] reference sequences)
-
- HGVS Nomenclature for the Description of Sequence Variants, http://www.hgvs.org/mutnomen/
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for RTT and MECP2)
References
-
- Rett A (1966) Uber ein eigenartiges hirnatrophisches Syndrom bei Hyperammonamie im Kindesalter. Wien Med Wochenschr 116:723–738 - PubMed
-
- Carney RM, Wolpert CM, Ravan SA, Shahbazian M, Ashley-Koch A, Cuccaro ML, Vance JM, Pericak-Vance MA (2003) Identification of MeCP2 mutations in a series of females with autistic disorder. Pediatr Neurol 28:205–211 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
