

Simulation of Ca-calmodulin-dependent protein kinase II on rabbit ventricular myocyte ion currents and action potentials
- PMID: 17704163
- PMCID: PMC2084250
- DOI: 10.1529/biophysj.107.114868
Simulation of Ca-calmodulin-dependent protein kinase II on rabbit ventricular myocyte ion currents and action potentials
Abstract
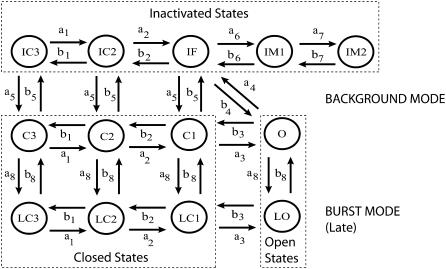
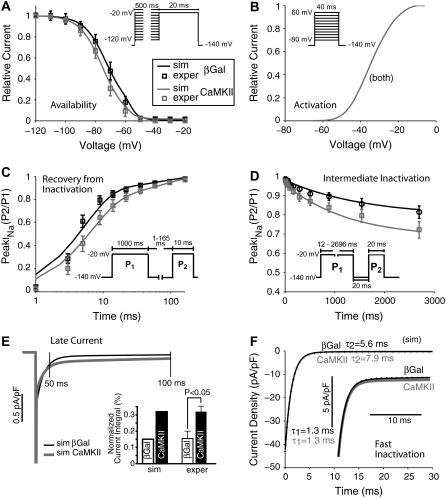
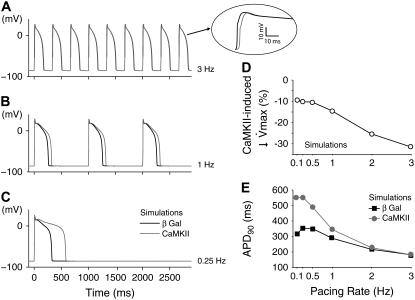
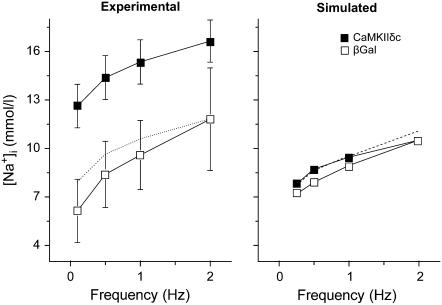
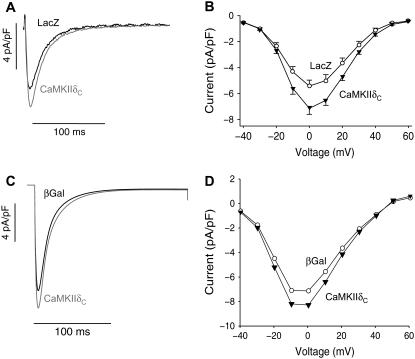
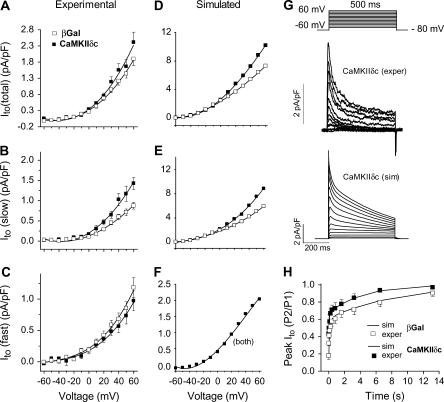
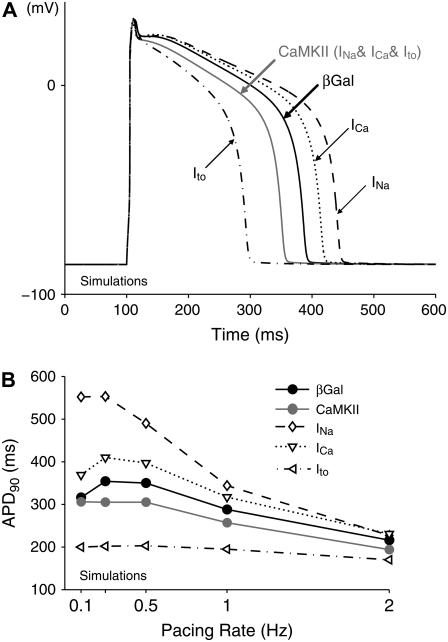
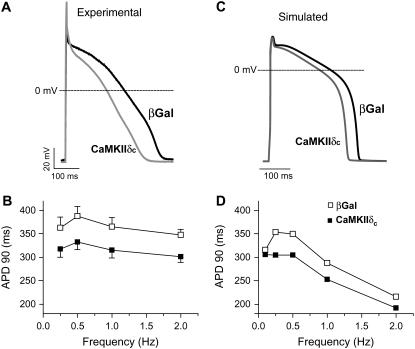
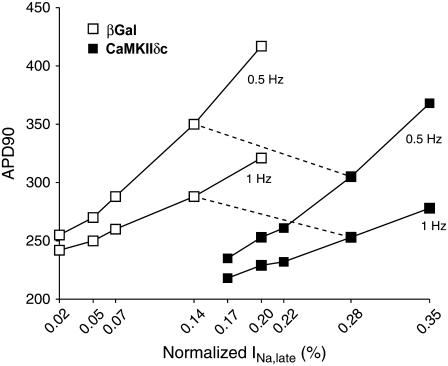
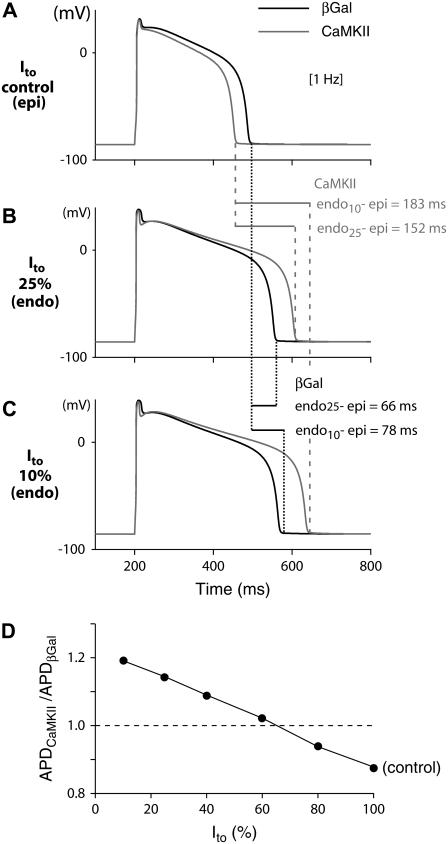
Ca-calmodulin-dependent protein kinase II (CaMKII) was recently shown to alter Na(+) channel gating and recapitulate a human Na(+) channel genetic mutation that causes an unusual combined arrhythmogenic phenotype in patients: simultaneous long QT syndrome and Brugada syndrome. CaMKII is upregulated in heart failure where arrhythmias are common, and CaMKII inhibition can reduce arrhythmias. Thus, CaMKII-dependent channel modulation may contribute to acquired arrhythmic disease. We developed a Markovian Na(+) channel model including CaMKII-dependent changes, and incorporated it into a comprehensive myocyte action potential (AP) model with Na(+) and Ca(2+) transport. CaMKII shifts Na(+) current (I(Na)) availability to more negative voltage, enhances intermediate inactivation, and slows recovery from inactivation (all loss-of-function effects), but also enhances late noninactivating I(Na) (gain of function). At slow heart rates, with long diastolic time for I(Na) recovery, late I(Na) is the predominant effect, leading to AP prolongation (long QT syndrome). At fast heart rates, where recovery time is limited and APs are shorter, there is little effect on AP duration, but reduced availability decreases I(Na), AP upstroke velocity, and conduction (Brugada syndrome). CaMKII also increases cardiac Ca(2+) and K(+) currents (I(Ca) and I(to)), complicating CaMKII-dependent AP changes. Incorporating I(Ca) and I(to) effects individually prolongs and shortens AP duration. Combining I(Na), I(Ca), and I(to) effects results in shortening of AP duration with CaMKII. With transmural heterogeneity of I(to) and I(to) downregulation in heart failure, CaMKII may accentuate dispersion of repolarization. This provides a useful initial framework to consider pathways by which CaMKII may contribute to arrhythmogenesis.
Figures
References
-
- Tan, H. 2006. Sodium channel variants in heart disease: expanding horizons. J. Cardiovasc. Electrophysiol. 17(Suppl 1):S151–S157. - PubMed
-
- Veldkamp, M. W., P. C. Viswanathan, C. Bezzina, A. Baartscheer, A. A. M. Wilde, and J. R. Balser. 2000. Two distinct congenital arrhythmias evoked by a multidysfunctional Na+ channel. Circ. Res. 86:e91–e97. - PubMed
-
- Ai, X., J. W. Curran, T. R. Shannon, D. M. Bers, and S. M. Pogwizd. 2005. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 97:1314–1322. - PubMed
-
- Maier, L. S., and D. M. Bers. 2007. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc. Res. 73:631–640. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
