

Elucidating the altered transcriptional programs in breast cancer using independent component analysis
- PMID: 17708679
- PMCID: PMC1950343
- DOI: 10.1371/journal.pcbi.0030161
Elucidating the altered transcriptional programs in breast cancer using independent component analysis
Abstract
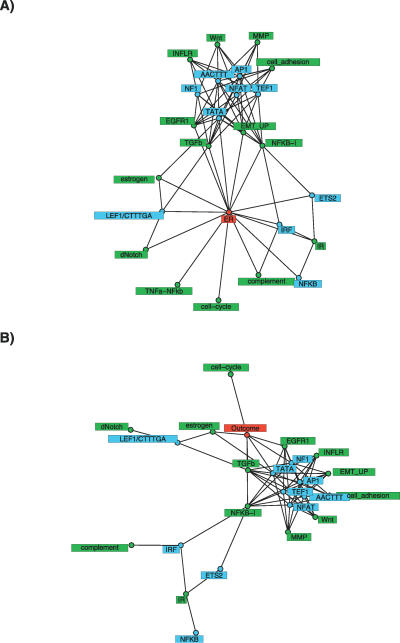
The quantity of mRNA transcripts in a cell is determined by a complex interplay of cooperative and counteracting biological processes. Independent Component Analysis (ICA) is one of a few number of unsupervised algorithms that have been applied to microarray gene expression data in an attempt to understand phenotype differences in terms of changes in the activation/inhibition patterns of biological pathways. While the ICA model has been shown to outperform other linear representations of the data such as Principal Components Analysis (PCA), a validation using explicit pathway and regulatory element information has not yet been performed. We apply a range of popular ICA algorithms to six of the largest microarray cancer datasets and use pathway-knowledge and regulatory-element databases for validation. We show that ICA outperforms PCA and clustering-based methods in that ICA components map closer to known cancer-related pathways, regulatory modules, and cancer phenotypes. Furthermore, we identify cancer signalling and oncogenic pathways and regulatory modules that play a prominent role in breast cancer and relate the differential activation patterns of these to breast cancer phenotypes. Importantly, we find novel associations linking immune response and epithelial-mesenchymal transition pathways with estrogen receptor status and histological grade, respectively. In addition, we find associations linking the activity levels of biological pathways and transcription factors (NF1 and NFAT) with clinical outcome in breast cancer. ICA provides a framework for a more biologically relevant interpretation of genomewide transcriptomic data. Adopting ICA as the analysis tool of choice will help understand the phenotype-pathway relationship and thus help elucidate the molecular taxonomy of heterogeneous cancers and of other complex genetic diseases.
Conflict of interest statement
Figures







References
-
- Stransky N, Vallot C, Reyal F, Bernard-Pierrot I, de Medina SG, et al. Regional copy number–independent deregulation of transcription in cancer. Nat Genet. 2006;38:1386–1396. - PubMed
-
- Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Barrette TR, Ghosh D, et al. Mining for regulatory programs in the cancer transcriptome. Nat Genet. 2005;37:579–583. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
