

Genome bioinformatic analysis of nonsynonymous SNPs
- PMID: 17708757
- PMCID: PMC1978506
- DOI: 10.1186/1471-2105-8-301
Genome bioinformatic analysis of nonsynonymous SNPs
Abstract
Background: Genome-wide association studies of common diseases for common, low penetrance causal variants are underway. A proportion of these will alter protein sequences, the most common of which is the non-synonymous single nucleotide polymorphism (nsSNP). It would be an advantage if the functional effects of an nsSNP on protein structure and function could be predicted, both for the final identification process of a causal variant in a disease-associated chromosome region, and in further functional analyses of the nsSNP and its disease-associated protein.
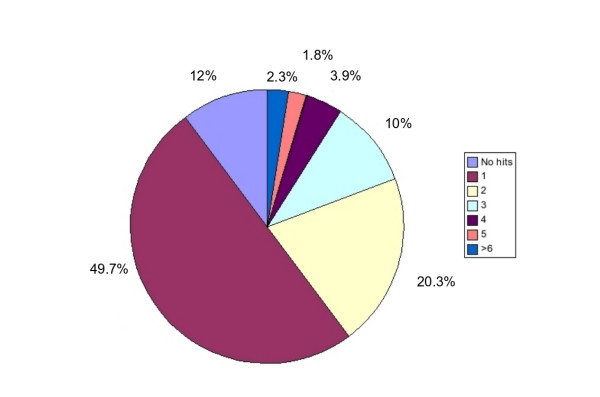
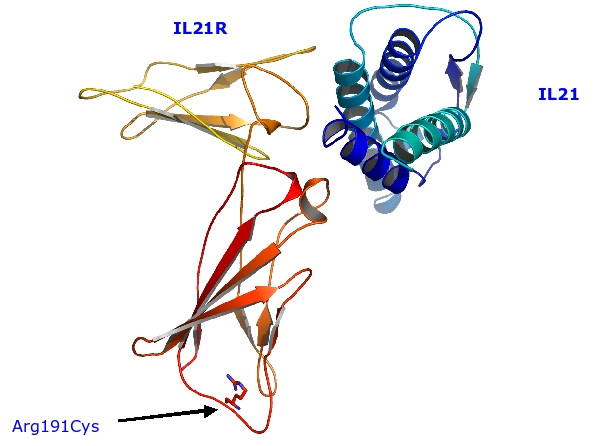
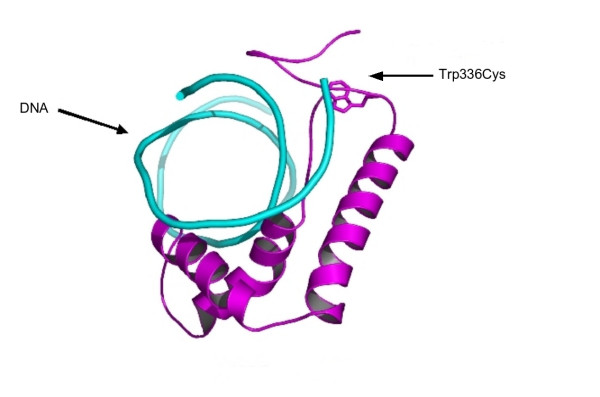
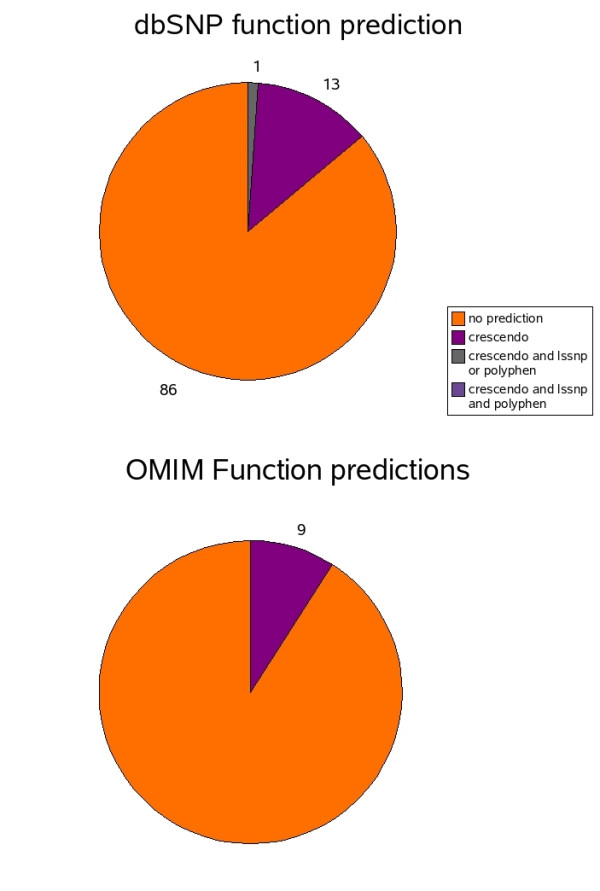
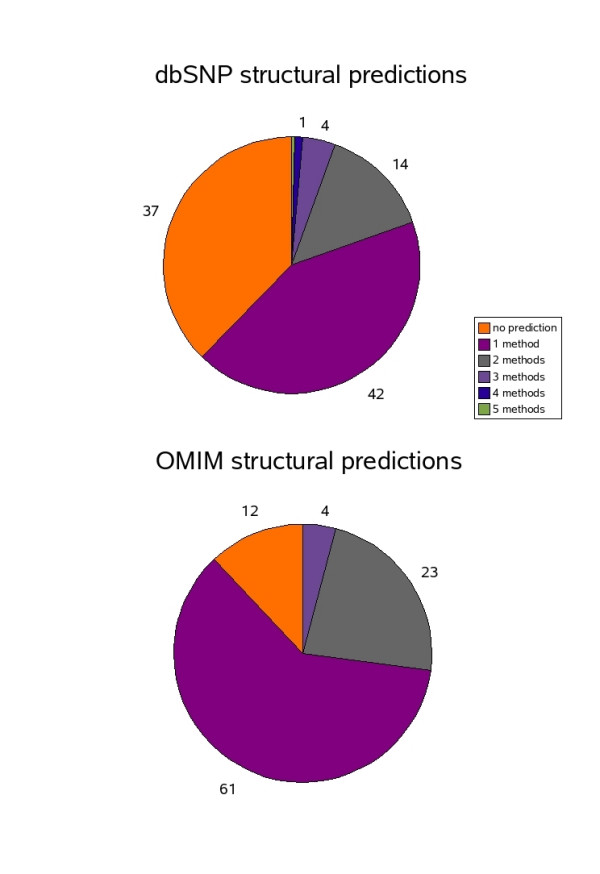
Results: In the present report we have compared and contrasted structure- and sequence-based methods of prediction to over 5500 genes carrying nearly 24,000 nsSNPs, by employing an automatic comparative modelling procedure to build models for the genes. The nsSNP information came from two sources, the OMIM database which are rare (minor allele frequency, MAF, < 0.01) and are known to cause penetrant, monogenic diseases. Secondly, nsSNP information came from dbSNP125, for which the vast majority of nsSNPs, mostly MAF > 0.05, have no known link to a disease. For over 40% of the nsSNPs, structure-based methods predicted which of these sequence changes are likely to either disrupt the structure of the protein or interfere with the function or interactions of the protein. For the remaining 60%, we generated sequence-based predictions.
Conclusion: We show that, in general, the prediction tools are able distinguish disease causing mutations from those mutations which are thought to have a neutral affect. We give examples of mutations in genes that are predicted to be deleterious and may have a role in disease. Contrary to previous reports, we also show that rare mutations are consistently predicted to be deleterious as often as commonly occurring nsSNPs.
Figures
References
-
- Collins FS, Brooks LD, Chakravarti A. A DNA Polymorphism Discovery Resource for Research on Human Genetic Variation. Genome Research. 1998;8:1229–1231. - PubMed
-
- The Hapmap database http://www.hapmap.org
-
- Clayton DG, Walker NM, Smyth DJ, Pask R, Cooper JD, Maier LM, Smink LJ, Lam AC, Ovington NR, Stevens HE, Nutland S, Howson JM, Faham M, Moorhead M, Jones HB, Falkowski M, Hardenbol P, Willis TD, Todd JA. Population structure, differential bias and genomic control in a large-scale, case-control association study. Nat Genet. 2005;37:1243–6. doi: 10.1038/ng1653. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
