

A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis
- PMID: 17710231
- PMCID: PMC1940239
- DOI: 10.1172/JCI31330
A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis
Abstract
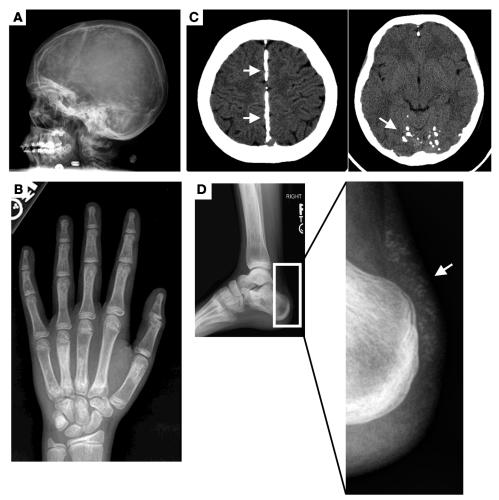
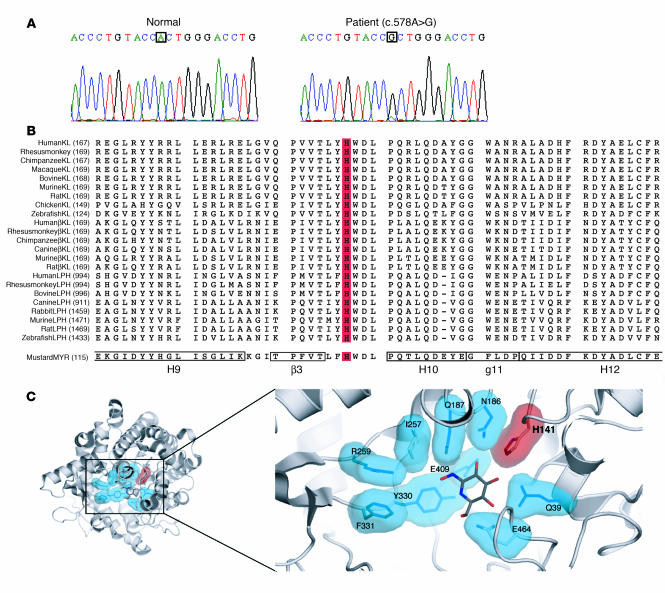
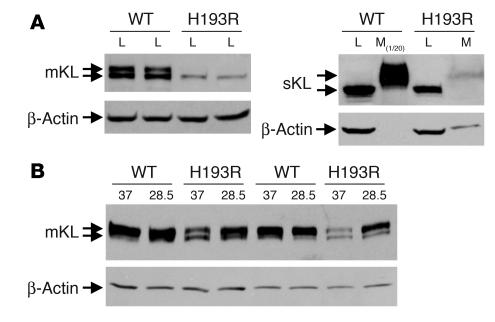
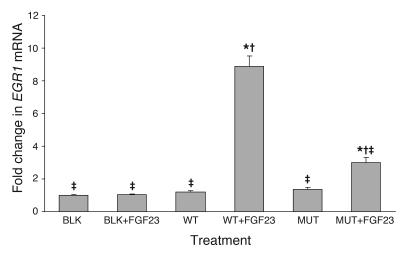
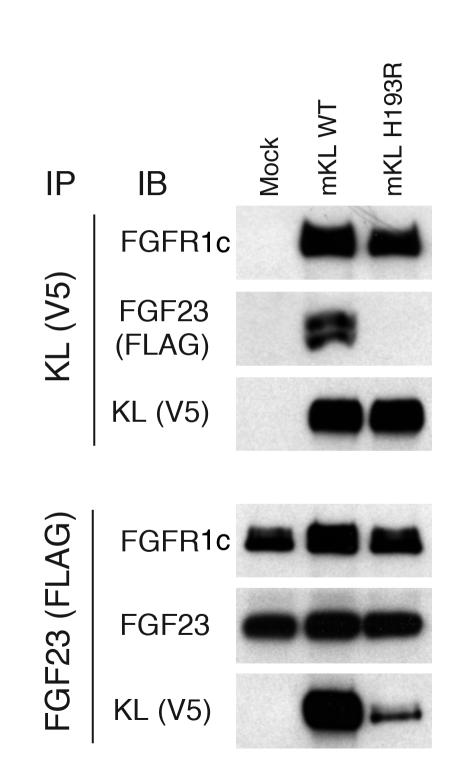
Familial tumoral calcinosis is characterized by ectopic calcifications and hyperphosphatemia due to inactivating mutations in FGF23 or UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 3 (GALNT3). Herein we report a homozygous missense mutation (H193R) in the KLOTHO (KL) gene of a 13-year-old girl who presented with severe tumoral calcinosis with dural and carotid artery calcifications. This patient exhibited defects in mineral ion homeostasis with marked hyperphosphatemia and hypercalcemia as well as elevated serum levels of parathyroid hormone and FGF23. Mapping of H193R mutation onto the crystal structure of myrosinase, a plant homolog of KL, revealed that this histidine residue was at the base of the deep catalytic cleft and mutation of this histidine to arginine should destabilize the putative glycosidase domain (KL1) of KL, thereby attenuating production of membrane-bound and secreted KL. Indeed, compared with wild-type KL, expression and secretion of H193R KL were markedly reduced in vitro, resulting in diminished ability of FGF23 to signal via its cognate FGF receptors. Taken together, our findings provide what we believe to be the first evidence that loss-of-function mutations in human KL impair FGF23 bioactivity, underscoring the essential role of KL in FGF23-mediated phosphate and vitamin D homeostasis in humans.
Figures
Similar articles
-
Ablation of the Galnt3 gene leads to low-circulating intact fibroblast growth factor 23 (Fgf23) concentrations and hyperphosphatemia despite increased Fgf23 expression.Endocrinology. 2009 Jun;150(6):2543-50. doi: 10.1210/en.2008-0877. Epub 2009 Feb 12. Endocrinology. 2009. PMID: 19213845 Free PMC article.
-
Familial tumoral calcinosis caused by a novel FGF23 mutation: response to induction of tubular renal acidosis with acetazolamide and the non-calcium phosphate binder sevelamer.Horm Res. 2009;71(3):178-84. doi: 10.1159/000197876. Epub 2009 Feb 3. Horm Res. 2009. PMID: 19188744
-
The role of mutant UDP-N-acetyl-alpha-D-galactosamine-polypeptide N-acetylgalactosaminyltransferase 3 in regulating serum intact fibroblast growth factor 23 and matrix extracellular phosphoglycoprotein in heritable tumoral calcinosis.J Clin Endocrinol Metab. 2006 Oct;91(10):4037-42. doi: 10.1210/jc.2006-0305. Epub 2006 Jul 25. J Clin Endocrinol Metab. 2006. PMID: 16868048
-
Congenital Hyperphosphatemic Conditions Caused by the Deficient Activity of FGF23.Calcif Tissue Int. 2021 Jan;108(1):104-115. doi: 10.1007/s00223-020-00659-6. Epub 2020 Jan 22. Calcif Tissue Int. 2021. PMID: 31965220 Review.
-
Hyperphosphatemic familial tumoral calcinosis: genetic models of deficient FGF23 action.Curr Osteoporos Rep. 2015 Apr;13(2):78-87. doi: 10.1007/s11914-015-0254-3. Curr Osteoporos Rep. 2015. PMID: 25656441 Review.
Cited by
-
Genetic disorders of phosphate regulation.Pediatr Nephrol. 2012 Sep;27(9):1477-87. doi: 10.1007/s00467-012-2103-2. Epub 2012 Feb 14. Pediatr Nephrol. 2012. PMID: 22350303 Free PMC article. Review.
-
Hyperphosphatemic Tumoral Calcinosis With Pemigatinib Use.AACE Clin Case Rep. 2022 Jul 16;8(5):217-220. doi: 10.1016/j.aace.2022.07.001. eCollection 2022 Sep-Oct. AACE Clin Case Rep. 2022. PMID: 36189136 Free PMC article.
-
MicroRNA-339 and microRNA-556 regulate Klotho expression in vitro.Age (Dordr). 2014 Feb;36(1):141-9. doi: 10.1007/s11357-013-9555-6. Epub 2013 Jul 2. Age (Dordr). 2014. PMID: 23818104 Free PMC article.
-
Interaction of Vitamin D with Peptide Hormones with Emphasis on Parathyroid Hormone, FGF23, and the Renin-Angiotensin-Aldosterone System.Nutrients. 2022 Dec 6;14(23):5186. doi: 10.3390/nu14235186. Nutrients. 2022. PMID: 36501215 Free PMC article. Review.
-
Circulating levels of soluble klotho and FGF23 in X-linked hypophosphatemia: circadian variance, effects of treatment, and relationship to parathyroid status.J Clin Endocrinol Metab. 2010 Nov;95(11):E352-7. doi: 10.1210/jc.2010-0589. Epub 2010 Aug 4. J Clin Endocrinol Metab. 2010. PMID: 20685863 Free PMC article.
References
-
- Araya K., et al. A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J. Clin. Endocrinol. Metab. 2005;90:5523–5527. - PubMed
-
- Benet-Pages A., Orlik P., Strom T.M., Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum. Mol. Genet. 2005;14:385–390. - PubMed
-
- Chefetz I., et al. A novel homozygous missense mutation in FGF23 causes Familial Tumoral Calcinosis associated with disseminated visceral calcification. Hum. Genet. 2005;118:261–266. - PubMed
-
- Larsson T., et al. A novel recessive mutation in fibroblast growth factor-23 causes familial tumoral calcinosis. J. Clin. Endocrinol. Metab. 2005;90:2424–2427. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
