

The Role of PPARs in Lung Fibrosis
- PMID: 17710235
- PMCID: PMC1940051
- DOI: 10.1155/2007/71323
The Role of PPARs in Lung Fibrosis
Abstract
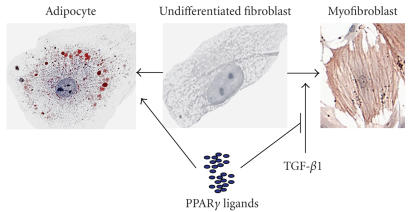
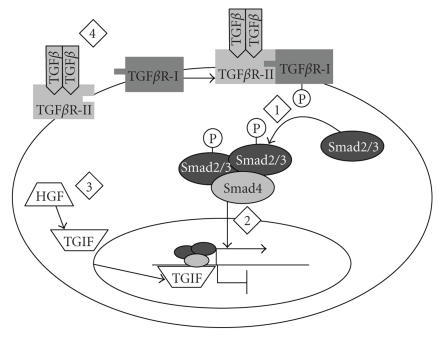
Pulmonary fibrosis is a group of disorders characterized by accumulation of scar tissue in the lung interstitium, resulting in loss of alveolar function, destruction of normal lung architecture, and respiratory distress. Some types of fibrosis respond to corticosteroids, but for many there are no effective treatments. Prognosis varies but can be poor. For example, patients with idiopathic pulmonary fibrosis (IPF) have a median survival of only 2.9 years. Prognosis may be better in patients with some other types of pulmonary fibrosis, and there is variability in survival even among individuals with biopsy-proven IPF. Evidence is accumulating that the peroxisome proliferator-activated receptors (PPARs) play important roles in regulating processes related to fibrogenesis, including cellular differentiation, inflammation, and wound healing. PPARalpha agonists, including the hypolidipemic fibrate drugs, inhibit the production of collagen by hepatic stellate cells and inhibit liver, kidney, and cardiac fibrosis in animal models. In the mouse model of lung fibrosis induced by bleomycin, a PPARalpha agonist significantly inhibited the fibrotic response, while PPARalpha knockout mice developed more serious fibrosis. PPARbeta/delta appears to play a critical role in regulating the transition from inflammation to wound healing. PPARbeta/delta agonists inhibit lung fibroblast proliferation and enhance the antifibrotic properties of PPARgamma agonists. PPARgamma ligands oppose the profibrotic effect of TGF-beta, which induces differentiation of fibroblasts to myofibroblasts, a critical effector cell in fibrosis. PPARgamma ligands, including the thiazolidinedione class of antidiabetic drugs, effectively inhibit lung fibrosis in vitro and in animal models. The clinical availability of potent and selective PPARalpha and PPARgamma agonists should facilitate rapid development of successful treatment strategies based on current and ongoing research.
Figures

Similar articles
-
The antifibrogenic potential of PPARgamma ligands in pulmonary fibrosis.J Investig Med. 2008 Feb;56(2):534-8. doi: 10.2310/JIM.0b013e31816464e9. J Investig Med. 2008. PMID: 18317437 Review.
-
Organ fibrosis inhibited by blocking transforming growth factor-β signaling via peroxisome proliferator-activated receptor γ agonists.Hepatobiliary Pancreat Dis Int. 2012 Oct;11(5):467-78. doi: 10.1016/s1499-3872(12)60210-0. Hepatobiliary Pancreat Dis Int. 2012. PMID: 23060391 Review.
-
PPARs and angiogenesis.Biochem Soc Trans. 2011 Dec;39(6):1601-5. doi: 10.1042/BST20110643. Biochem Soc Trans. 2011. PMID: 22103494 Review.
-
Peroxisome proliferator-activated receptors (PPARs): novel therapeutic targets in renal disease.Kidney Int. 2001 Jul;60(1):14-30. doi: 10.1046/j.1523-1755.2001.00766.x. Kidney Int. 2001. PMID: 11422732 Review.
-
Compromised peroxisomes in idiopathic pulmonary fibrosis, a vicious cycle inducing a higher fibrotic response via TGF-β signaling.Proc Natl Acad Sci U S A. 2015 Apr 21;112(16):E2048-57. doi: 10.1073/pnas.1415111112. Epub 2015 Apr 6. Proc Natl Acad Sci U S A. 2015. PMID: 25848047 Free PMC article.
Cited by
-
Lipid Mediators Regulate Pulmonary Fibrosis: Potential Mechanisms and Signaling Pathways.Int J Mol Sci. 2020 Jun 15;21(12):4257. doi: 10.3390/ijms21124257. Int J Mol Sci. 2020. PMID: 32549377 Free PMC article. Review.
-
PPARγ as a Potential Target to Treat Airway Mucus Hypersecretion in Chronic Airway Inflammatory Diseases.PPAR Res. 2012;2012:256874. doi: 10.1155/2012/256874. Epub 2012 Jun 17. PPAR Res. 2012. PMID: 22761606 Free PMC article.
-
ssRNA Virus and Host Lipid Rearrangements: Is There a Role for Lipid Droplets in SARS-CoV-2 Infection?Front Mol Biosci. 2020 Oct 8;7:578964. doi: 10.3389/fmolb.2020.578964. eCollection 2020. Front Mol Biosci. 2020. PMID: 33134318 Free PMC article. Review.
-
Beyond epithelial damage: vascular and endothelial contributions to idiopathic pulmonary fibrosis.J Clin Invest. 2023 Sep 15;133(18):e172058. doi: 10.1172/JCI172058. J Clin Invest. 2023. PMID: 37712420 Free PMC article. Review.
-
Asiatic acid prevents renal fibrosis in UUO rats via promoting the production of 15d-PGJ2, an endogenous ligand of PPAR-γ.Acta Pharmacol Sin. 2020 Mar;41(3):373-382. doi: 10.1038/s41401-019-0319-4. Epub 2019 Nov 8. Acta Pharmacol Sin. 2020. PMID: 31705123 Free PMC article.
References
-
- Thannickal VJ, Toews GB, White ES, Lynch JP, III, Martinez FJ. Mechanisms of pulmonary fibrosis. Annual Review of Medicine. 2004;55:395–417. - PubMed
-
- Sime PJ, O'Reilly KMA. Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clinical Immunology. 2001;99(3):308–319. - PubMed
-
- Geiser T. Idiopathic pulmonary fibrosis—a disorder of alveolar wound repair? Swiss Medical Weekly. 2003;133(29-30):405–411. - PubMed
-
- Fujimura N. Pathology and pathophysiology of pneumoconiosis. Current Opinion in Pulmonary Medicine. 2000;6(2):140–144. - PubMed
-
- Mossman BT, Churg A. Mechanisms in the pathogenesis of asbestosis and silicosis. American Journal of Respiratory and Critical Care Medicine. 1998;157(5, part 1):1666–1680. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
