

The RAGNYA fold: a novel fold with multiple topological variants found in functionally diverse nucleic acid, nucleotide and peptide-binding proteins
- PMID: 17715145
- PMCID: PMC2034487
- DOI: 10.1093/nar/gkm558
The RAGNYA fold: a novel fold with multiple topological variants found in functionally diverse nucleic acid, nucleotide and peptide-binding proteins
Abstract
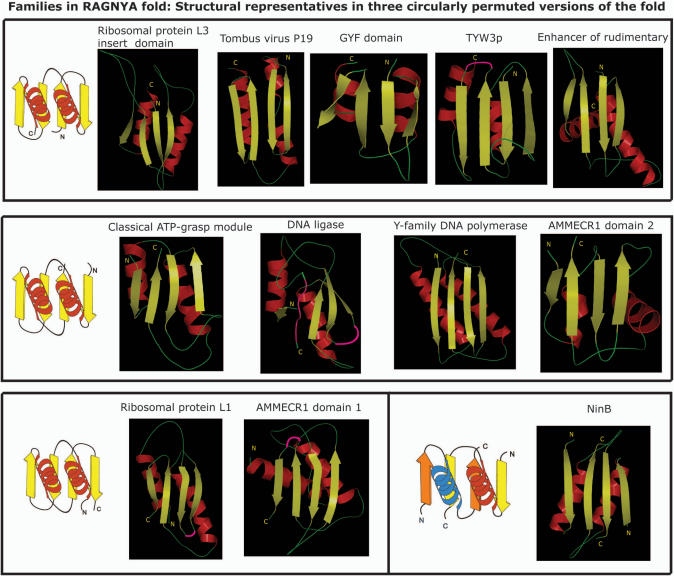
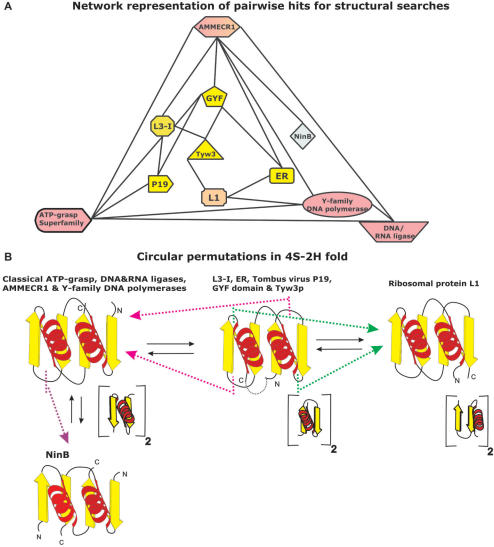
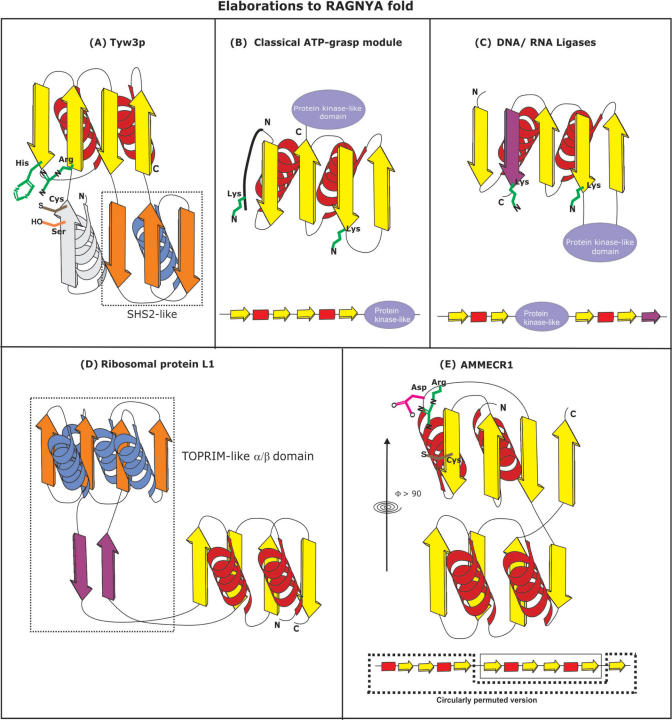
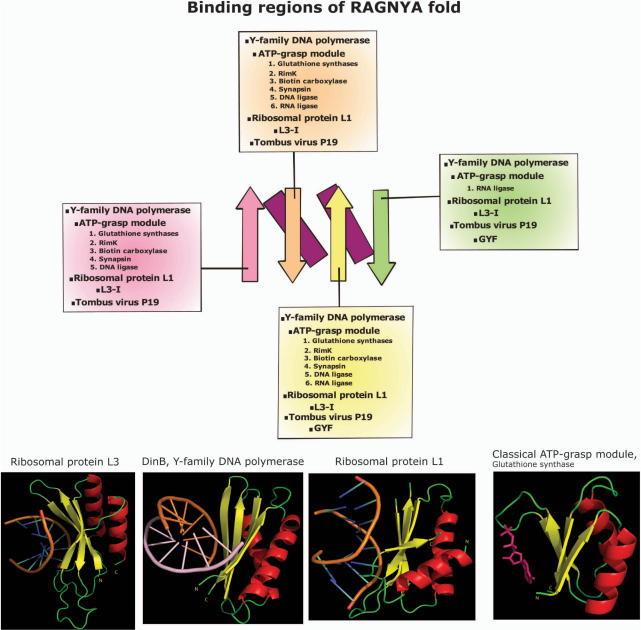
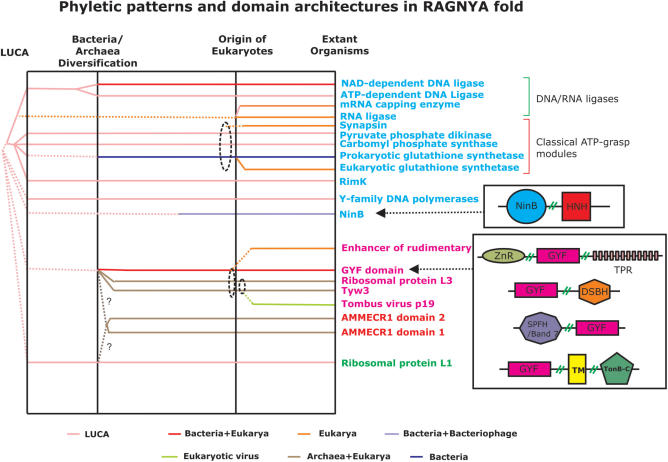
Using sensitive structure similarity searches, we identify a shared alpha+beta fold, RAGNYA, principally involved in nucleic acid, nucleotide or peptide interactions in a diverse group of proteins. These include the Ribosomal proteins L3 and L1, ATP-grasp modules, the GYF domain, DNA-recombination proteins of the NinB family from caudate bacteriophages, the C-terminal DNA-interacting domain of the Y-family DNA polymerases, the uncharacterized enzyme AMMECR1, the siRNA silencing repressor of tombusviruses, tRNA Wybutosine biosynthesis enzyme Tyw3p, DNA/RNA ligases and related nucleotidyltransferases and the Enhancer of rudimentary proteins. This fold exhibits three distinct circularly permuted versions and is composed of an internal repeat of a unit with two-strands and a helix. We show that despite considerable structural diversity in the fold, its representatives show a common mode of nucleic acid or nucleotide interaction via the exposed face of the sheet. Using this information and sensitive profile-based sequence searches: (1) we predict the active site, and mode of substrate interaction of the Wybutosine biosynthesis enzyme, Tyw3p, and a potential catalytic role for AMMECR1. (2) We provide insights regarding the mode of nucleic acid interaction of the NinB proteins, and the evolution of the active site of classical ATP-grasp enzymes and DNA/RNA ligases. (3) We also present evidence for a bacterial origin of the GYF domain and propose how this version of the fold might have been utilized in peptide interactions in the context of nucleoprotein complexes.
Figures
Similar articles
-
The emergence of catalytic and structural diversity within the beta-clip fold.Proteins. 2004 Jun 1;55(4):977-91. doi: 10.1002/prot.20076. Proteins. 2004. PMID: 15146494
-
Diversification of catalytic activities and ligand interactions in the protein fold shared by the sugar isomerases, eIF2B, DeoR transcription factors, acyl-CoA transferases and methenyltetrahydrofolate synthetase.J Mol Biol. 2006 Feb 24;356(3):823-42. doi: 10.1016/j.jmb.2005.11.031. Epub 2005 Dec 1. J Mol Biol. 2006. PMID: 16376935
-
Novel conserved domains in proteins with predicted roles in eukaryotic cell-cycle regulation, decapping and RNA stability.BMC Genomics. 2004 Jul 16;5(1):45. doi: 10.1186/1471-2164-5-45. BMC Genomics. 2004. PMID: 15257761 Free PMC article.
-
Nucleic acid recognition by OB-fold proteins.Annu Rev Biophys Biomol Struct. 2003;32:115-33. doi: 10.1146/annurev.biophys.32.110601.142506. Epub 2003 Feb 18. Annu Rev Biophys Biomol Struct. 2003. PMID: 12598368 Free PMC article. Review.
-
The polynucleotide ligase and RNA capping enzyme superfamily of covalent nucleotidyltransferases.Curr Opin Struct Biol. 2004 Dec;14(6):757-64. doi: 10.1016/j.sbi.2004.10.006. Curr Opin Struct Biol. 2004. PMID: 15582400 Review.
Cited by
-
Early Evolution of Transcription Systems and Divergence of Archaea and Bacteria.Front Mol Biosci. 2021 May 5;8:651134. doi: 10.3389/fmolb.2021.651134. eCollection 2021. Front Mol Biosci. 2021. PMID: 34026831 Free PMC article. Review.
-
Apprehending the NAD+-ADPr-Dependent Systems in the Virus World.Viruses. 2022 Sep 7;14(9):1977. doi: 10.3390/v14091977. Viruses. 2022. PMID: 36146784 Free PMC article.
-
RNA damage in biological conflicts and the diversity of responding RNA repair systems.Nucleic Acids Res. 2016 Oct 14;44(18):8525-8555. doi: 10.1093/nar/gkw722. Epub 2016 Aug 17. Nucleic Acids Res. 2016. PMID: 27536007 Free PMC article. Review.
-
The enigmatic ERH protein: its role in cell cycle, RNA splicing and cancer.Protein Cell. 2013 Nov;4(11):807-12. doi: 10.1007/s13238-013-3056-3. Protein Cell. 2013. PMID: 24078386 Free PMC article. Review.
-
Insights from the architecture of the bacterial transcription apparatus.J Struct Biol. 2012 Sep;179(3):299-319. doi: 10.1016/j.jsb.2011.12.013. Epub 2011 Dec 24. J Struct Biol. 2012. PMID: 22210308 Free PMC article.
References
-
- Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. - PubMed
-
- Aravind L, Anantharaman V, Balaji S, Babu MM, Iyer LM. The many faces of the helix-turn-helix domain: transcription regulation and beyond. FEMS Microbiol. Rev. 2005;29:231–262. - PubMed
-
- Murzin AG. A ribosomal protein module in EF-G and DNA gyrase. Nat. Struct. Biol. 1995;2:25–26. - PubMed
