

Molecular evolution of pathogenicity-island genes in Pseudomonas viridiflava
- PMID: 17720907
- PMCID: PMC2034611
- DOI: 10.1534/genetics.107.077925
Molecular evolution of pathogenicity-island genes in Pseudomonas viridiflava
Abstract
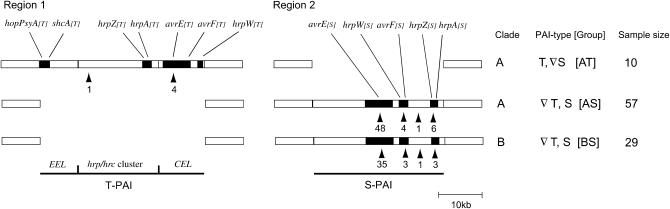
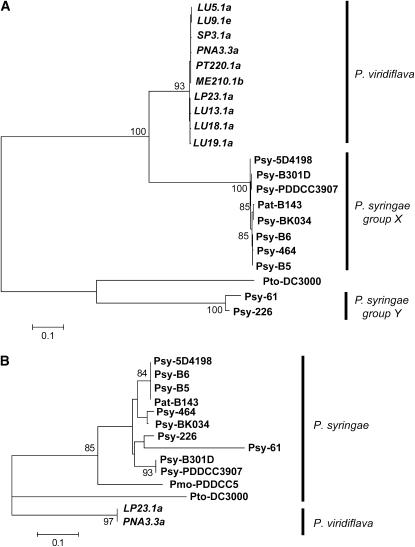
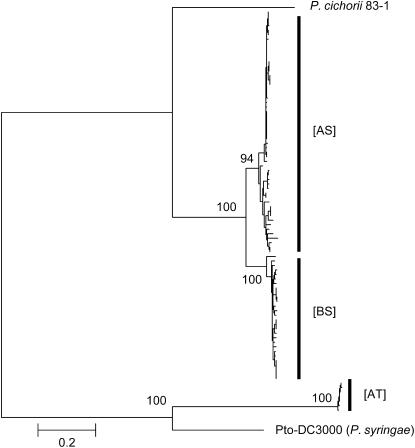
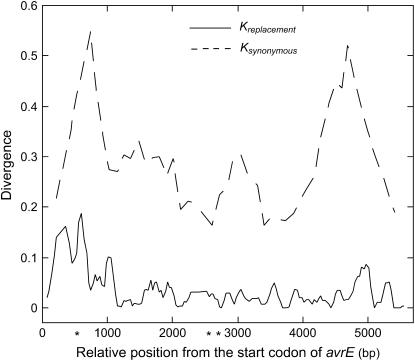
The bacterial pathogen Pseudomonas viridiflava possesses two pathogenicity islands (PAIs) that share many gene homologs, but are structurally and phenotypically differentiated (T-PAI and S-PAI). These PAIs are paralogous, but only one is present in each isolate. While this dual presence/absence polymorphism has been shown to be maintained by balancing selection, little is known about the molecular evolution of individual genes on the PAIs. Here we investigate genetic variation of 12 PAI gene loci (7 on T-PAI and 5 on S-PAI) in 96 worldwide isolates of P. viridiflava. These genes include avirulence genes (hopPsyA and avrE), their putative chaperones (shcA and avrF), and genes encoding the type III outer proteins (hrpA, hrpZ, and hrpW). Average nucleotide diversities in these genes (pi = 0.004-0.020) were close to those in the genetic background. Large numbers of recombination events were found within PAIs and a sign of positive selection was detected in avrE. These results suggest that the PAI genes are evolving relatively freely from each other on the PAIs, rather than as a single unit under balancing selection. Evolutionarily stable PAIs may be preferable in this species because preexisting genetic variation enables P. viridiflava to respond rapidly to natural selection.
Figures
References
-
- Alfano, J. R., A. O. Charkowski, W. L. Deng, J. L. Badel, T. Petnicki-Ocwieja et al., 2000. The Pseudomonas syringae Hrp pathogenicity island has a tripartite mosaic structure composed of a cluster of type III secretion genes bounded by exchangeable effector and conserved effector loci that contribute to parasitic fitness and pathogenicity in plants. Proc. Natl. Acad. Sci. USA 97: 4856–4861. - PMC - PubMed
-
- Arabidopsis Genome Initiative, 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815. - PubMed
-
- Arnold, D. L., R. W. Jackson, A. J. Fillingham, S. C. Goss, J. D. Taylor et al., 2001. Highly conserved sequences flank avirulence genes: isolation of novel avirulence genes from Pseudomonas syringae pv. pisi. Microbiology 147: 1171–1182. - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
