

Acute downregulation of ENaC by EGF involves the PY motif and putative ERK phosphorylation site
- PMID: 17724164
- PMCID: PMC2151644
- DOI: 10.1085/jgp.200709775
Acute downregulation of ENaC by EGF involves the PY motif and putative ERK phosphorylation site
Abstract
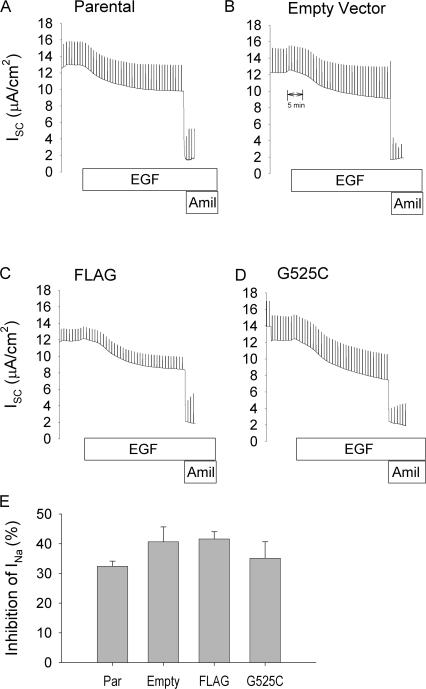
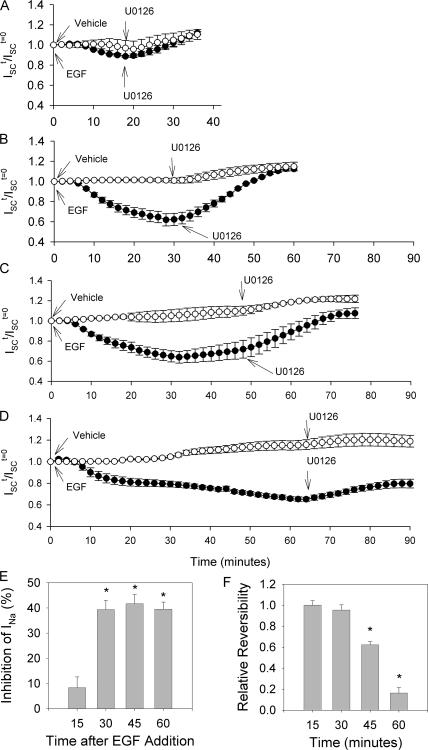
The epithelial sodium channel (ENaC) is expressed in a variety of tissues, including the renal collecting duct, where it constitutes the rate-limiting step for sodium reabsorption. Liddle's syndrome is caused by gain-of-function mutations in the beta and gamma subunits of ENaC, resulting in enhanced Na reabsorption and hypertension. Epidermal growth factor (EGF) causes acute inhibition of Na absorption in collecting duct principal cells via an extracellular signal-regulated kinase (ERK)-dependent mechanism. In experiments with primary cultures of collecting duct cells derived from a mouse model of Liddle's disease (beta-ENaC truncation), it was found that EGF inhibited short-circuit current (Isc) by 24 +/- 5% in wild-type cells but only by 6 +/- 3% in homozygous mutant cells. In order to elucidate the role of specific regions of the beta-ENaC C terminus, Madin-Darby canine kidney (MDCK) cell lines that express beta-ENaC with mutation of the PY motif (P616L), the ERK phosphorylation site (T613A), and C terminus truncation (R564stop) were created using the Phoenix retroviral system. All three mutants exhibited significant attenuation of the EGF-induced inhibition of sodium current. In MDCK cells with wild-type beta-ENaC, EGF-induced inhibition of Isc (<30 min) was fully reversed by exposure to an ERK kinase inhibitor and occurred with no change in ENaC surface expression, indicative of an effect on channel open probability (P(o)). At later times (>30 min), EGF-induced inhibition of Isc was not reversed by an ERK kinase inhibitor and was accompanied by a decrease in ENaC surface expression. Our results are consistent with an ERK-mediated decrease in ENaC open probability and enhanced retrieval of sodium channels from the apical membrane.
Figures










Similar articles
-
Epithelial Na+ channel mutants causing Liddle's syndrome retain ability to respond to aldosterone and vasopressin.Am J Physiol Renal Physiol. 2003 Sep;285(3):F459-71. doi: 10.1152/ajprenal.00071.2003. Epub 2003 May 20. Am J Physiol Renal Physiol. 2003. PMID: 12759227
-
EGF and its related growth factors mediate sodium transport in mpkCCDc14 cells via ErbB2 (neu/HER-2) receptor.J Cell Physiol. 2010 Apr;223(1):252-9. doi: 10.1002/jcp.22033. J Cell Physiol. 2010. PMID: 20049896
-
Abnormal EGF-dependent regulation of sodium absorption in ARPKD collecting duct cells.Am J Physiol Renal Physiol. 2005 Mar;288(3):F474-82. doi: 10.1152/ajprenal.00227.2004. Epub 2004 Nov 2. Am J Physiol Renal Physiol. 2005. PMID: 15522985
-
Regulation of the epithelial Na+ channel by Nedd4 and ubiquitination.Kidney Int. 2000 Mar;57(3):809-15. doi: 10.1046/j.1523-1755.2000.00919.x. Kidney Int. 2000. PMID: 10720933 Review.
-
Regulation of epithelial sodium channels by the ubiquitin-proteasome proteolytic pathway.Am J Physiol Renal Physiol. 2006 Jun;290(6):F1285-94. doi: 10.1152/ajprenal.00432.2005. Am J Physiol Renal Physiol. 2006. PMID: 16682484 Review.
Cited by
-
Regulation of the epithelial Na+ channel by the protein kinase CK2.J Biol Chem. 2008 May 9;283(19):13225-32. doi: 10.1074/jbc.M704532200. Epub 2008 Feb 28. J Biol Chem. 2008. PMID: 18308722 Free PMC article.
-
Phosphoproteomic response to epidermal growth factor in native rat inner medullary collecting duct.Am J Physiol Renal Physiol. 2025 Jan 1;328(1):F29-F47. doi: 10.1152/ajprenal.00182.2024. Epub 2024 Nov 7. Am J Physiol Renal Physiol. 2025. PMID: 39508840
-
Regulation of the epithelial sodium channel by membrane trafficking.Am J Physiol Renal Physiol. 2009 Jan;296(1):F10-24. doi: 10.1152/ajprenal.90248.2008. Epub 2008 May 28. Am J Physiol Renal Physiol. 2009. PMID: 18508877 Free PMC article. Review.
-
Regulation of sodium transport by ENaC in the kidney.Curr Opin Nephrol Hypertens. 2010 Jan;19(1):98-105. doi: 10.1097/MNH.0b013e328332bda4. Curr Opin Nephrol Hypertens. 2010. PMID: 19996890 Free PMC article. Review.
-
Nerve growth factor reduces amiloride-sensitive Na+ transport in human airway epithelial cells.Physiol Rep. 2014 Jul 16;2(7):e12073. doi: 10.14814/phy2.12073. Physiol Rep. 2014. PMID: 25347857 Free PMC article.
References
-
- Auberson, M., N. Hoffmann-Pochon, A. Vandewalle, S. Kellenberger, and L. Schild. 2003. Epithelial Na+ channel mutants causing Liddle's syndrome retain ability to respond to aldosterone and vasopressin. Am. J. Physiol. Renal Physiol. 285:F459–F471. - PubMed
-
- Blazer-Yost, B.L., X. Liu, and S.I. Helman. 1998. Hormonal regulation of ENaCs: insulin and aldosterone. Am. J. Physiol. 274:C1373–C1379. - PubMed
-
- Booth, R.E., and J.D. Stockand. 2003. Targeted degradation of ENaC in response to PKC activation of the ERK1/2 cascade. Am. J. Physiol. Renal Physiol. 284:F938–F947. - PubMed
-
- Bruns, J.B., M.D. Carattino, S. Sheng, A.B. Maarouf, O.A. Weisz, J.M. Pilewski, R.P. Hughey, T.R. Kleyman. 2007. Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the γ subunit. J. Biol. Chem. 282:6153–6160. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
