

Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration
- PMID: 17726112
- PMCID: PMC1964831
- DOI: 10.1073/pnas.0701311104
Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration
Abstract
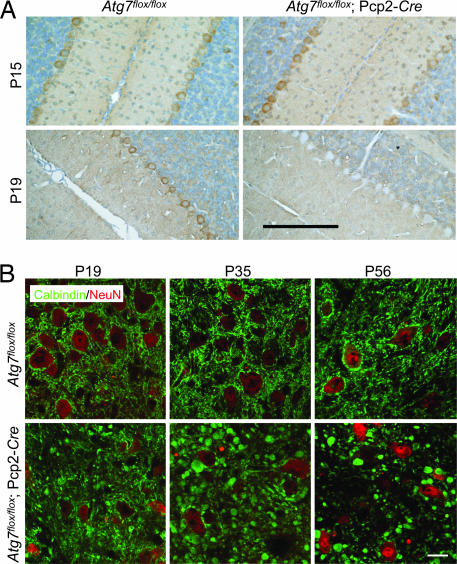
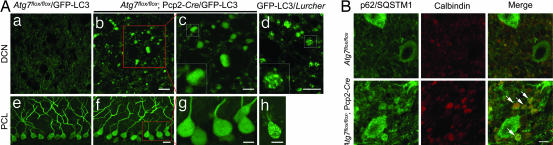
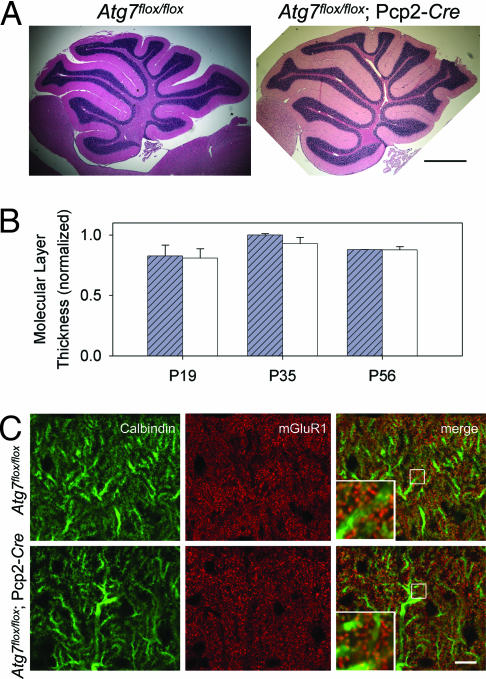
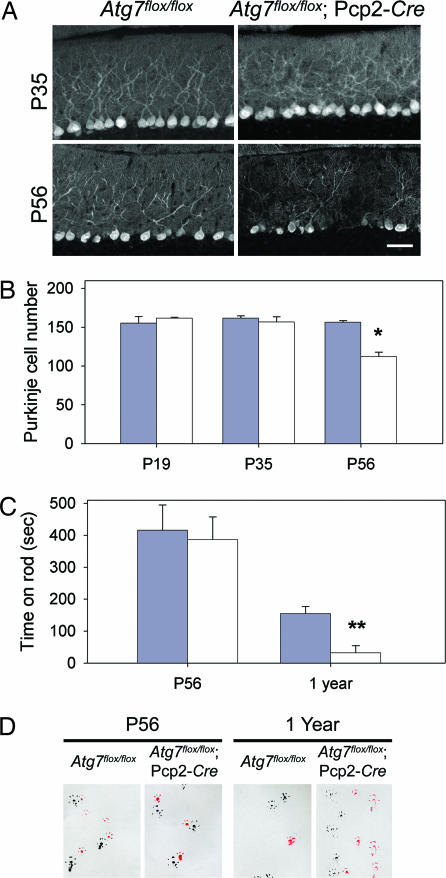
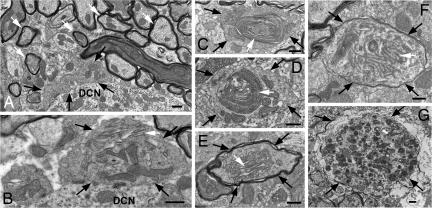
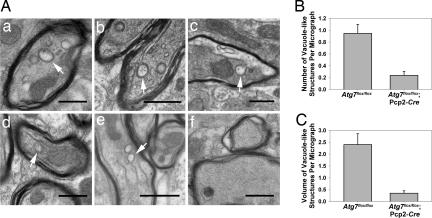
Autophagy is a regulated lysosomal degradation process that involves autophagosome formation and transport. Although recent evidence indicates that basal levels of autophagy protect against neurodegeneration, the exact mechanism whereby this occurs is not known. By using conditional knockout mutant mice, we report that neuronal autophagy is particularly important for the maintenance of local homeostasis of axon terminals and protection against axonal degeneration. We show that specific ablation of an essential autophagy gene, Atg7, in Purkinje cells initially causes cell-autonomous, progressive dystrophy (manifested by axonal swellings) and degeneration of the axon terminals. Consistent with suppression of autophagy, no autophagosomes are observed in these dystrophic swellings, which is in contrast to accumulation of autophagosomes in the axonal dystrophic swellings under pathological conditions. Axonal dystrophy of mutant Purkinje cells proceeds with little sign of dendritic or spine atrophy, indicating that axon terminals are much more vulnerable to autophagy impairment than dendrites. This early pathological event in the axons is followed by cell-autonomous Purkinje cell death and mouse behavioral deficits. Furthermore, ultrastructural analyses of mutant Purkinje cells reveal an accumulation of aberrant membrane structures in the axonal dystrophic swellings. Finally, we observe double-membrane vacuole-like structures in wild-type Purkinje cell axons, whereas these structures are abolished in mutant Purkinje cell axons. Thus, we conclude that the autophagy protein Atg7 is required for membrane trafficking and turnover in the axons. Our study implicates impairment of axonal autophagy as a possible mechanism for axonopathy associated with neurodegeneration.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Levine B, Klionsky DJ. Dev Cell. 2004;6:463–477. - PubMed
-
- Rubinszstein DC, DiFiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B, Stefanis L, Tolkovsky A. Autophagy. 2005;1:11–22. - PubMed
-
- Yue Z, Horton A, Bravin M, DeJager PL, Selimi F, Heintz N. Neuron. 2002;35:921–933. - PubMed
-
- Dixon JS. Nature. 1967;215:657–658. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
