

NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence
- PMID: 17726526
- PMCID: PMC1949492
- DOI: 10.1371/journal.pone.0000796
NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence
Abstract
Background: Binding of peptides to Major Histocompatibility Complex (MHC) molecules is the single most selective step in the recognition of pathogens by the cellular immune system. The human MHC class I system (HLA-I) is extremely polymorphic. The number of registered HLA-I molecules has now surpassed 1500. Characterizing the specificity of each separately would be a major undertaking.
Principal findings: Here, we have drawn on a large database of known peptide-HLA-I interactions to develop a bioinformatics method, which takes both peptide and HLA sequence information into account, and generates quantitative predictions of the affinity of any peptide-HLA-I interaction. Prospective experimental validation of peptides predicted to bind to previously untested HLA-I molecules, cross-validation, and retrospective prediction of known HIV immune epitopes and endogenous presented peptides, all successfully validate this method. We further demonstrate that the method can be applied to perform a clustering analysis of MHC specificities and suggest using this clustering to select particularly informative novel MHC molecules for future biochemical and functional analysis.
Conclusions: Encompassing all HLA molecules, this high-throughput computational method lends itself to epitope searches that are not only genome- and pathogen-wide, but also HLA-wide. Thus, it offers a truly global analysis of immune responses supporting rational development of vaccines and immunotherapy. It also promises to provide new basic insights into HLA structure-function relationships. The method is available at http://www.cbs.dtu.dk/services/NetMHCpan.
Conflict of interest statement
Figures

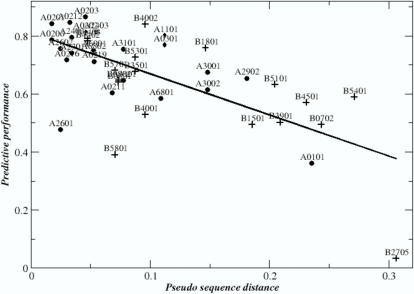
 , where s(A,B) is the BLOSUM50 alignment score between the pseudo sequences for alleles A and B, respectively. HLA-A alleles are shown as solid circles. HLA-B alleles are shown as +. The Pearson correlation coefficient between the pseudo sequence distance and the predictive performance for the 42 HLA alleles included in the plot is 0.67. Note, that the distance measure inherently assumes that all residues are equally important and independent of the pseudo sequence context. While this assumption is obviously inconsistent with the reality of primary anchors, it meets another essential requirement; it is simple and unbiased.
, where s(A,B) is the BLOSUM50 alignment score between the pseudo sequences for alleles A and B, respectively. HLA-A alleles are shown as solid circles. HLA-B alleles are shown as +. The Pearson correlation coefficient between the pseudo sequence distance and the predictive performance for the 42 HLA alleles included in the plot is 0.67. Note, that the distance measure inherently assumes that all residues are equally important and independent of the pseudo sequence context. While this assumption is obviously inconsistent with the reality of primary anchors, it meets another essential requirement; it is simple and unbiased.

References
-
- Lauemoller SL, Kesmir C, Corbet SL, Fomsgaard A, Holm A, et al. Identifying cytotoxic T cell epitopes from genomic and proteomic information: “The human MHC project.”. Rev Immunogenet. 2000;2:477–491. - PubMed
-
- Yewdell JW, Bennink JR. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annual Review of Immunology. 1999;17:51–88. - PubMed
-
- Sette A, Fikes J. Epitope-based vaccines: an update on epitope identification, vaccine design and delivery. Curr Opin Immunol. 2003;15:461–470. - PubMed
-
- Sette A, Sidney J. Nine major HLA class I supertypes account for the vast preponderance of HLA-A and –B polymorphism. Immunogenetics. 1999;50:201–212. - PubMed
-
- Lund O, Nielsen M, Kesmir C, Petersen AG, Lundegaard C, et al. Definition of supertypes for HLA molecules using clustering of specificity matrices. Immunogenetics. 2004;55:797–810. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
