

Contribution of exogenous genetic elements to the group A Streptococcus metagenome
- PMID: 17726530
- PMCID: PMC1949102
- DOI: 10.1371/journal.pone.0000800
Contribution of exogenous genetic elements to the group A Streptococcus metagenome
Abstract
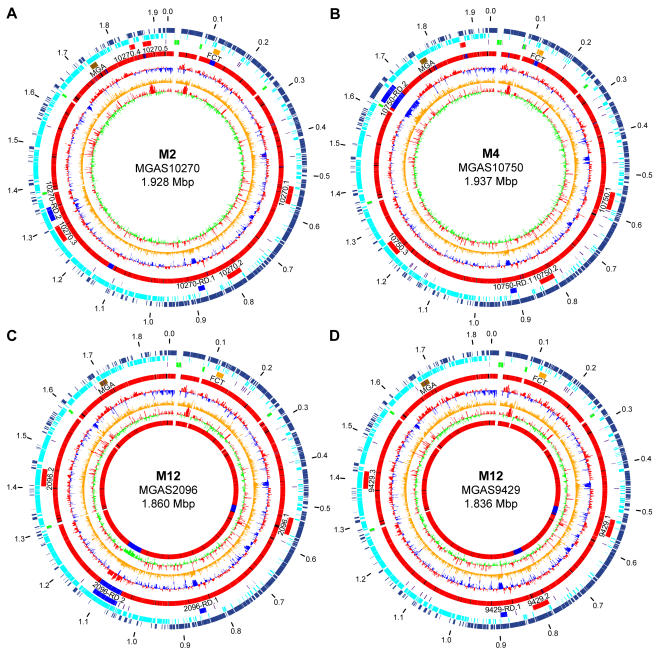
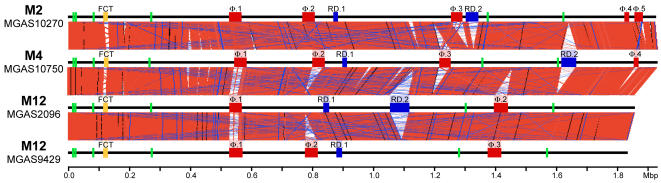
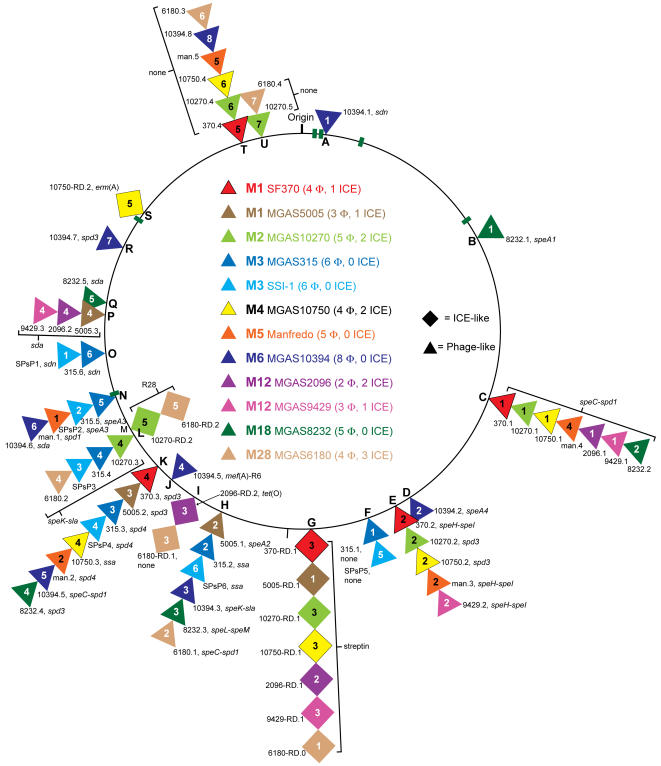
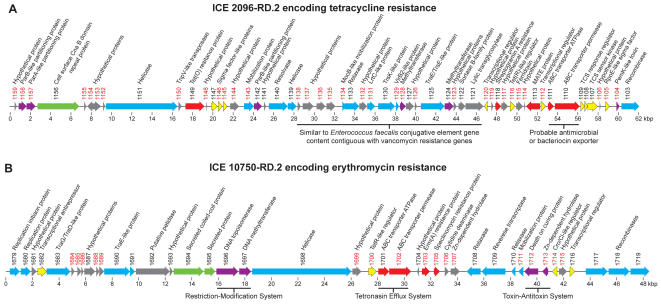
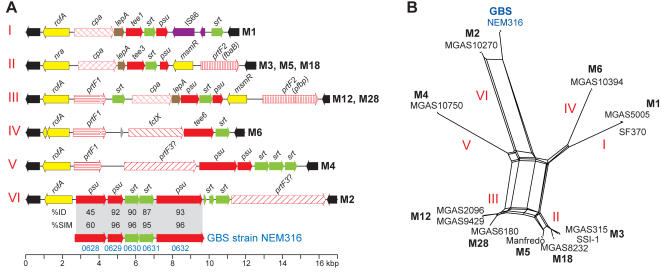
Variation in gene content among strains of a bacterial species contributes to biomedically relevant differences in phenotypes such as virulence and antimicrobial resistance. Group A Streptococcus (GAS) causes a diverse array of human infections and sequelae, and exhibits a complex pathogenic behavior. To enhance our understanding of genotype-phenotype relationships in this important pathogen, we determined the complete genome sequences of four GAS strains expressing M protein serotypes (M2, M4, and 2 M12) that commonly cause noninvasive and invasive infections. These sequences were compared with eight previously determined GAS genomes and regions of variably present gene content were assessed. Consistent with the previously determined genomes, each of the new genomes is approximately 1.9 Mb in size, with approximately 10% of the gene content of each encoded on variably present exogenous genetic elements. Like the other GAS genomes, these four genomes are polylysogenic and prophage encode the majority of the variably present gene content of each. In contrast to most of the previously determined genomes, multiple exogenous integrated conjugative elements (ICEs) with characteristics of conjugative transposons and plasmids are present in these new genomes. Cumulatively, 242 new GAS metagenome genes were identified that were not present in the previously sequenced genomes. Importantly, ICEs accounted for 41% of the new GAS metagenome gene content identified in these four genomes. Two large ICEs, designated 2096-RD.2 (63 kb) and 10750-RD.2 (49 kb), have multiple genes encoding resistance to antimicrobial agents, including tetracycline and erythromycin, respectively. Also resident on these ICEs are three genes encoding inferred extracellular proteins of unknown function, including a predicted cell surface protein that is only present in the genome of the serotype M12 strain cultured from a patient with acute poststreptococcal glomerulonephritis. The data provide new information about the GAS metagenome and will assist studies of pathogenesis, antimicrobial resistance, and population genomics.
Conflict of interest statement
Figures
References
-
- Fitzgerald JR, Musser JM. Evolutionary genomics of pathogenic bacteria. Trends Microbiol. 2001;9:547–553. - PubMed
-
- Glaser P, Rusniok C, Buchrieser C, Chevalier F, Frangeul L, et al. Genome sequence of Streptococcus agalactiae, a pathogen causing invasive neonatal disease. Mol Microbiol. 2002;45:1499–1513. - PubMed
-
- Lindsay JA, Holden MT. Staphylococcus aureus: superbug, super genome? Trends Microbiol. 2004;12:378–385. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
