

Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A
- PMID: 17728232
- PMCID: PMC2169025
- DOI: 10.1128/JVI.01369-07
Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A
Abstract
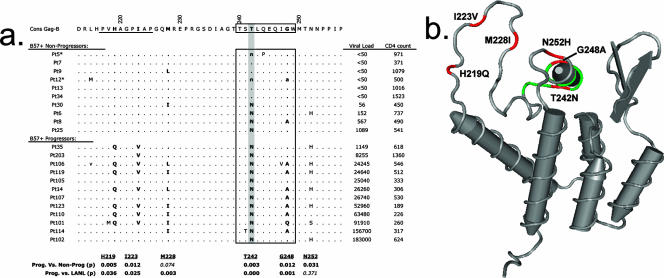
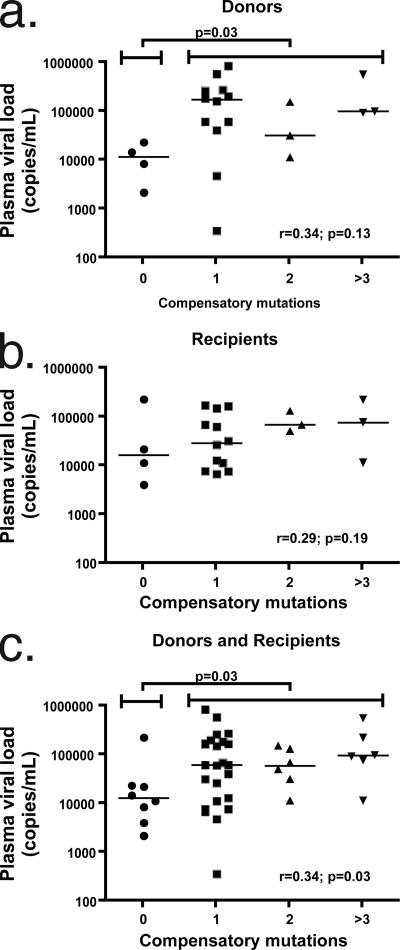
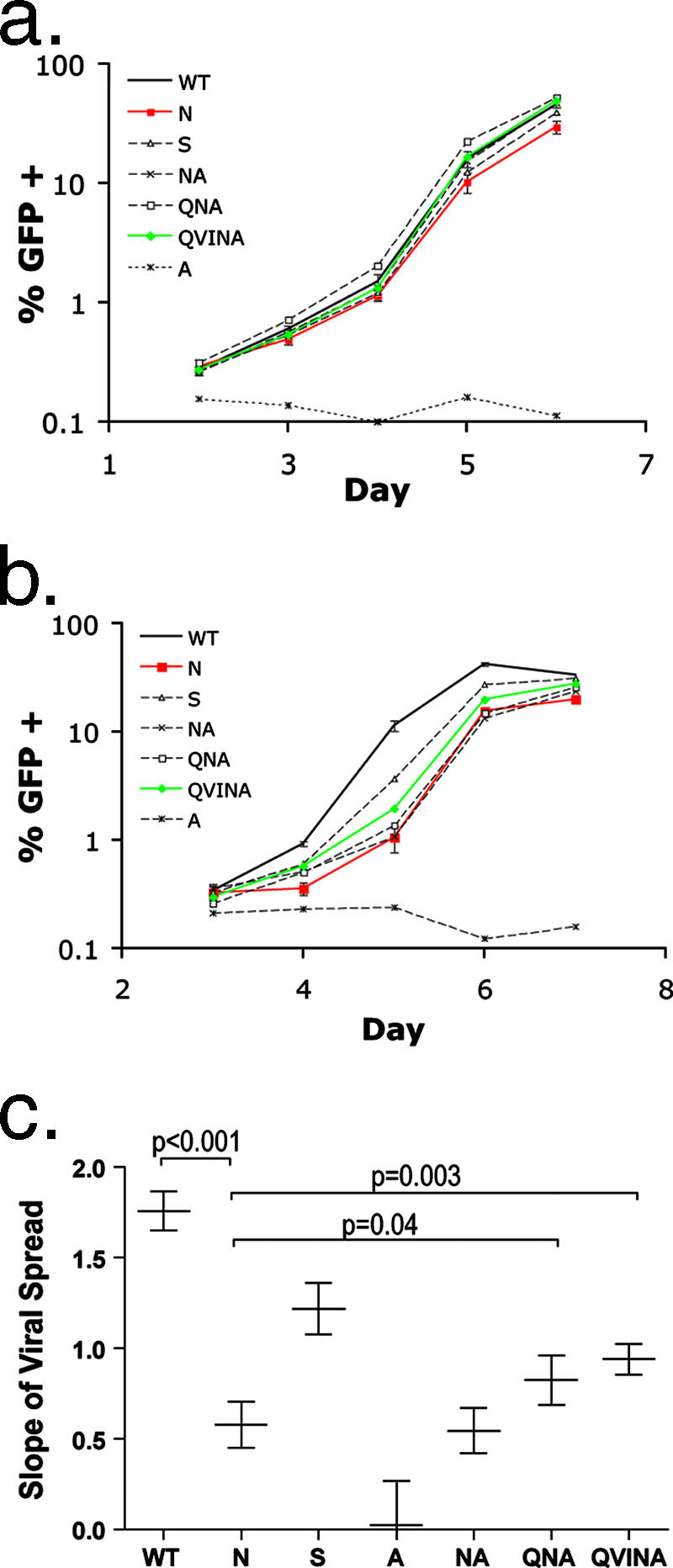
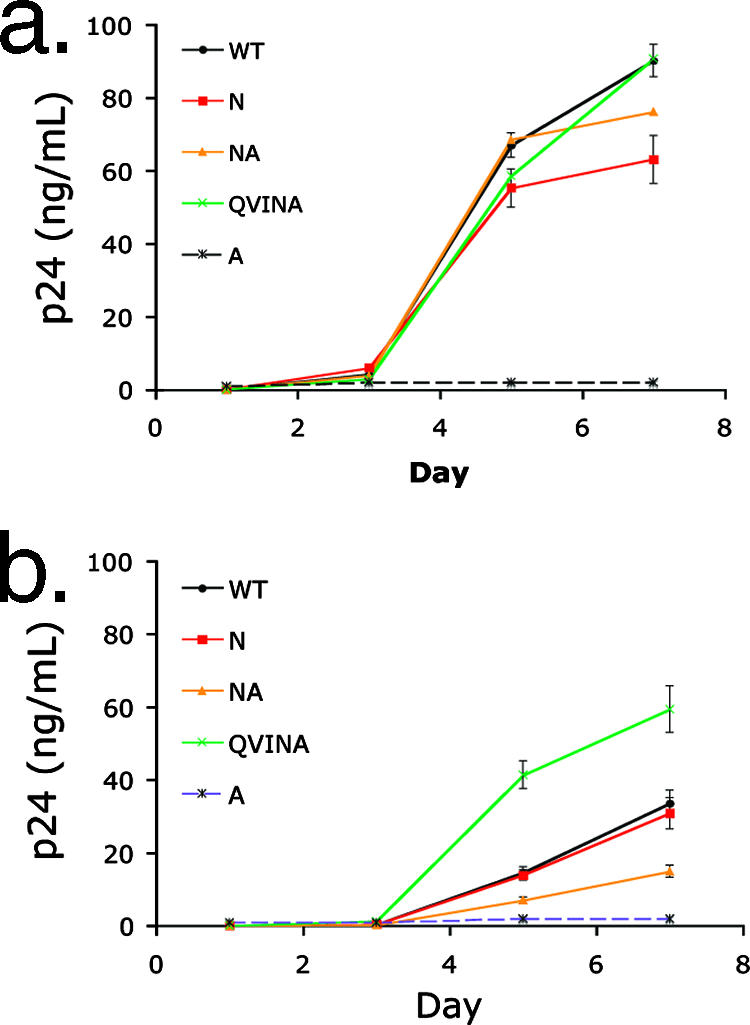
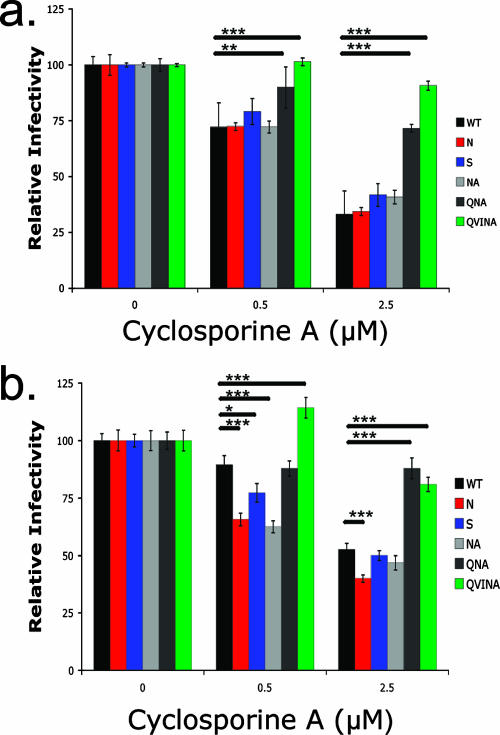
Certain histocompatibility leukocyte antigen (HLA) alleles are associated with improved clinical outcomes for individuals infected with human immunodeficiency virus type 1 (HIV-1), but the mechanisms for their effects remain undefined. An early CD8(+) T-cell escape mutation in the dominant HLA-B57-restricted Gag epitope TW10 (TSTLQEQIGW) has been shown to impair HIV-1 replication capacity in vitro. We demonstrate here that this T(242)N substitution in the capsid protein is associated with upstream mutations at residues H(219), I(223), and M(228) in the cyclophilin A (CypA)-binding loop in B57(+) individuals with progressive disease. In an independent cohort of epidemiologically linked transmission pairs, the presence of these substitutions in viruses encoding T(242)N was associated with significantly higher plasma viremia in donors, further suggesting that these secondary mutations compensated for the replication defect of T(242)N. Using NL4-3 constructs, we illustrate the ability of these CypA loop changes to partially restore replication of the T(242)N variant in vitro. Notably, these mutations also enhanced viral resistance to the drug cyclosporine A, indicating a reduced dependence of the compensated virus on CypA that is normally essential for optimal infectivity. Therefore, mutations in TW10 allow HIV-1 to evade a dominant early CD8(+) T-cell response, but the benefits of escape are offset by a defect in capsid function. These data suggest that TW10 escape variants undergo a postentry block that is partially overcome by changes in the CypA-binding loop and identify a mechanism for an HIV-1 fitness defect that may contribute to the slower disease progression associated with HLA-B57.
Figures

References
-
- Agarwal, P. K. 2004. Cis/trans isomerization in HIV-1 capsid protein catalyzed by cyclophilin A: insights from computational and theoretical studies. Proteins 56:449-463. - PubMed
-
- Allen, T. M., M. Altfeld, S. C. Geer, E. T. Kalife, C. Moore, M. O'Sullivan, K. I. Desouza, M. E. Feeney, R. L. Eldridge, E. L. Maier, D. E. Kaufmann, M. P. Lahaie, L. Reyor, G. Tanzi, M. N. Johnston, C. Brander, R. Draenert, J. K. Rockstroh, H. Jessen, E. S. Rosenberg, S. A. Mallal, and B. D. Walker. 2005. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. J. Virol. 79:13239-13249. - PMC - PubMed
-
- Allen, T. M., M. Altfeld, X. G. Yu, K. M. O'Sullivan, M. Lichterfeld, S. Le Gall, M. John, B. R. Mothe, P. K. Lee, E. T. Kalife, D. E. Cohen, K. A. Freedberg, D. A. Strick, M. N. Johnston, A. Sette, E. S. Rosenberg, S. A. Mallal, P. J. Goulder, C. Brander, and B. D. Walker. 2004. Selection, transmission, and reversion of an antigen-processing cytotoxic T-lymphocyte escape mutation in human immunodeficiency virus type 1 infection. J. Virol. 78:7069-7078. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
