

Contribution of cystine-glutamate antiporters to the psychotomimetic effects of phencyclidine
- PMID: 17728701
- PMCID: PMC3907109
- DOI: 10.1038/sj.npp.1301532
Contribution of cystine-glutamate antiporters to the psychotomimetic effects of phencyclidine
Abstract
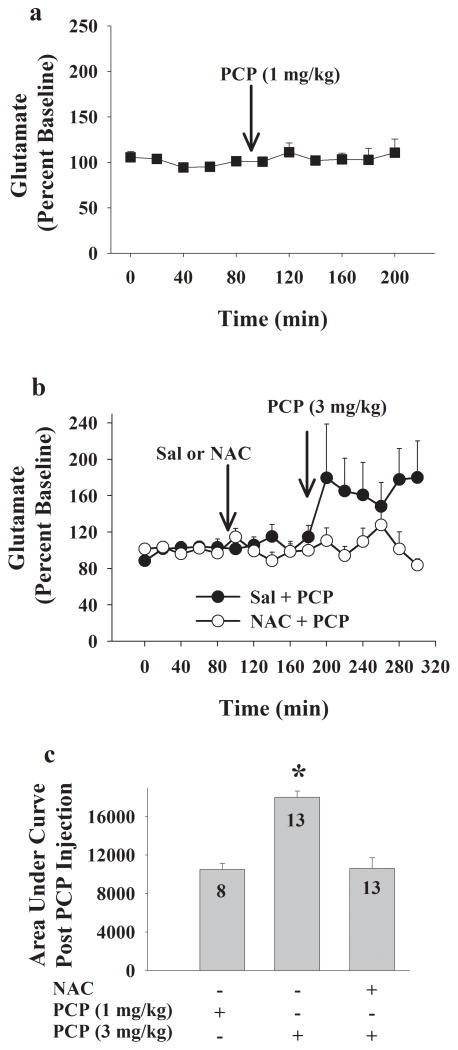
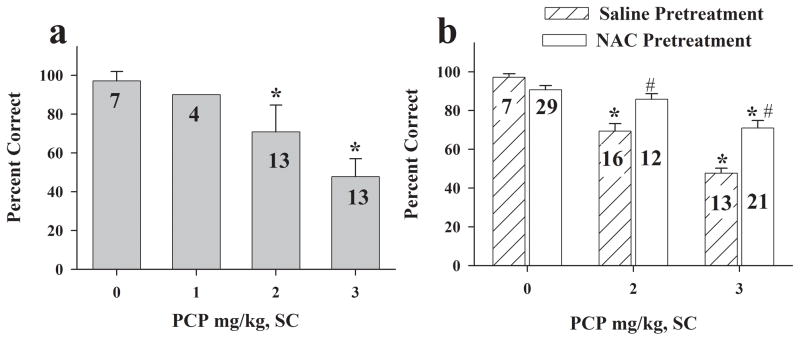
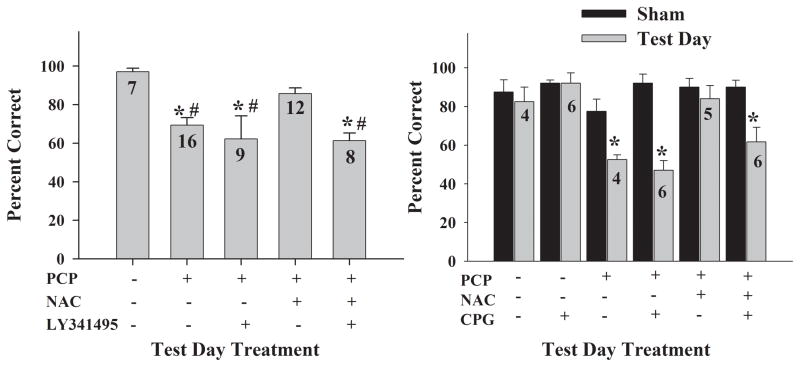
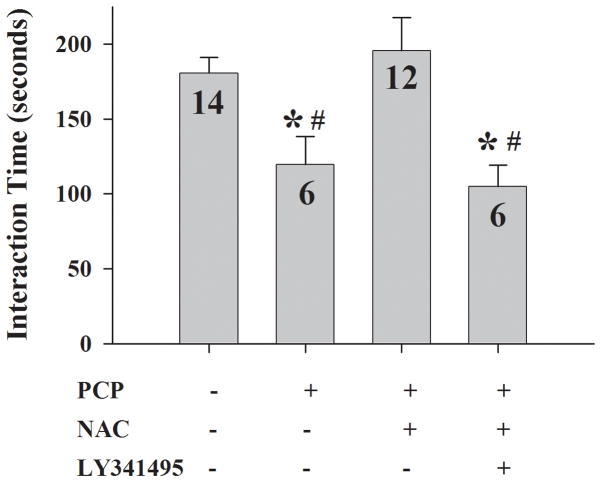
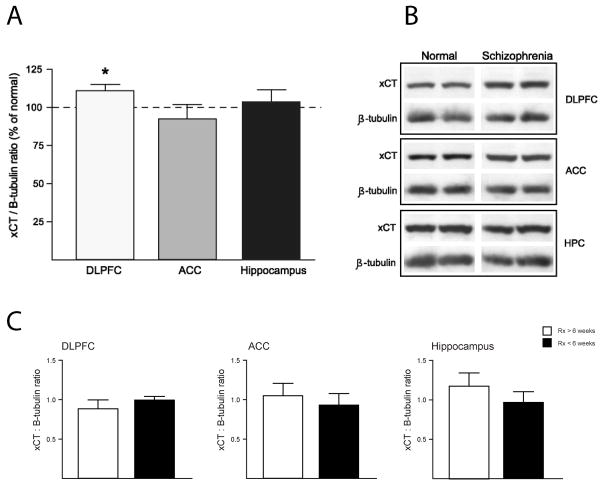
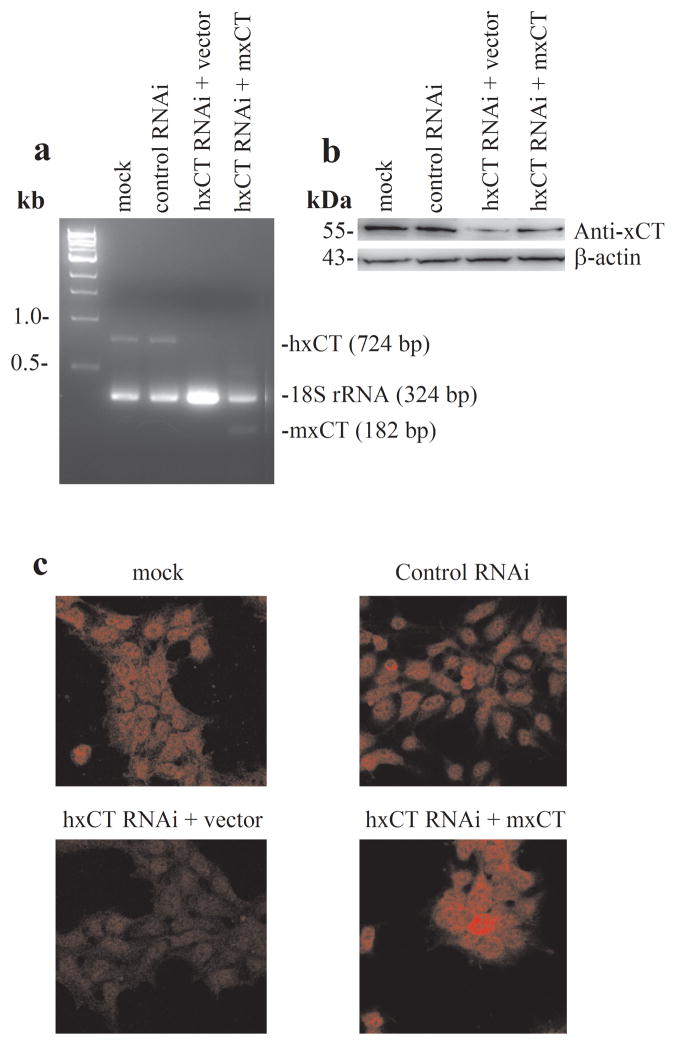
Altered glutamate signaling contributes to a myriad of neural disorders, including schizophrenia. While synaptic levels are intensely studied, nonvesicular release mechanisms, including cystine-glutamate exchange, maintain high steady-state glutamate levels in the extrasynaptic space. The existence of extrasynaptic receptors, including metabotropic group II glutamate receptors (mGluR), pose nonvesicular release mechanisms as unrecognized targets capable of contributing to pathological glutamate signaling. We tested the hypothesis that activation of cystine-glutamate antiporters using the cysteine prodrug N-acetylcysteine would blunt psychotomimetic effects in the rodent phencyclidine (PCP) model of schizophrenia. First, we demonstrate that PCP elevates extracellular glutamate in the prefrontal cortex, an effect that is blocked by N-acetylcysteine pretreatment. To determine the relevance of the above finding, we assessed social interaction and found that N-acetylcysteine reverses social withdrawal produced by repeated PCP. In a separate paradigm, acute PCP resulted in working memory deficits assessed using a discrete trial t-maze task, and this effect was also reversed by N-acetylcysteine pretreatment. The capacity of N-acetylcysteine to restore working memory was blocked by infusion of the cystine-glutamate antiporter inhibitor (S)-4-carboxyphenylglycine into the prefrontal cortex or systemic administration of the group II mGluR antagonist LY341495 indicating that the effects of N-acetylcysteine requires cystine-glutamate exchange and group II mGluR activation. Finally, protein levels from postmortem tissue obtained from schizophrenic patients revealed significant changes in the level of xCT, the active subunit for cystine-glutamate exchange, in the dorsolateral prefrontal cortex. These data advance cystine-glutamate antiporters as novel targets capable of reversing the psychotomimetic effects of PCP.
Conflict of interest statement
Dr. James Meador-Woodruff is the editor in chief of Neuropsychopharmacology and the ACNP website and receives a stipend from ACNP for these services. The remaining authors do not have a conflict of interest or any relationship with outside organizations to disclose.
Figures
Similar articles
-
N-acetylcysteine modulates hallucinogenic 5-HT(2A) receptor agonist-mediated responses: behavioral, molecular, and electrophysiological studies.Neuropharmacology. 2014 Jun;81:215-23. doi: 10.1016/j.neuropharm.2014.02.006. Epub 2014 Feb 15. Neuropharmacology. 2014. PMID: 24534112
-
Reduction in phencyclidine induced sensorimotor gating deficits in the rat following increased system xc⁻ activity in the medial prefrontal cortex.Psychopharmacology (Berl). 2013 Apr;226(3):531-40. doi: 10.1007/s00213-012-2926-3. Epub 2012 Nov 29. Psychopharmacology (Berl). 2013. PMID: 23192314 Free PMC article.
-
Effects of cariprazine on extracellular levels of glutamate, GABA, dopamine, noradrenaline and serotonin in the medial prefrontal cortex in the rat phencyclidine model of schizophrenia studied by microdialysis and simultaneous recordings of locomotor activity.Psychopharmacology (Berl). 2018 May;235(5):1593-1607. doi: 10.1007/s00213-018-4874-z. Epub 2018 Apr 11. Psychopharmacology (Berl). 2018. PMID: 29637288 Free PMC article.
-
Animal model of schizophrenia: dysfunction of NMDA receptor-signaling in mice following withdrawal from repeated administration of phencyclidine.Ann N Y Acad Sci. 2006 Nov;1086:160-8. doi: 10.1196/annals.1377.003. Ann N Y Acad Sci. 2006. PMID: 17185514 Review.
-
The role of the hippocampo-prefrontal cortex system in phencyclidine-induced psychosis: a model for schizophrenia.J Physiol Paris. 2013 Dec;107(6):434-40. doi: 10.1016/j.jphysparis.2013.06.002. Epub 2013 Jun 17. J Physiol Paris. 2013. PMID: 23792022 Review.
Cited by
-
Amantadine Combines Astroglial System Xc- Activation with Glutamate/NMDA Receptor Inhibition.Biomolecules. 2019 May 17;9(5):191. doi: 10.3390/biom9050191. Biomolecules. 2019. PMID: 31108896 Free PMC article.
-
Potential psychiatric applications of metabotropic glutamate receptor agonists and antagonists.CNS Drugs. 2010 Aug;24(8):669-93. doi: 10.2165/11533230-000000000-00000. CNS Drugs. 2010. PMID: 20658799 Review.
-
Overview on the Effects of N-Acetylcysteine in Neurodegenerative Diseases.Molecules. 2018 Dec 13;23(12):3305. doi: 10.3390/molecules23123305. Molecules. 2018. PMID: 30551603 Free PMC article. Review.
-
Effects of N-acetylcysteine on amphetamine-induced sensitization in mice.Braz J Psychiatry. 2017 Dec 11;40(2):169-173. doi: 10.1590/1516-4446-2017-2337. Print 2018 Apr-June. Braz J Psychiatry. 2017. PMID: 29236922 Free PMC article.
-
Implication of the glutamate-cystine antiporter xCT in schizophrenia cases linked to impaired GSH synthesis.NPJ Schizophr. 2017 Sep 18;3(1):31. doi: 10.1038/s41537-017-0035-3. NPJ Schizophr. 2017. PMID: 28924227 Free PMC article.
References
-
- Aoyama K, Suh SW, Hamby AM, Liu J, Chan WY, Chen Y, Swanson RA. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci. 2006;9:119–126. - PubMed
-
- Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, Kalivas PW. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci. 2003;6:743–749. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
