

Phylogenomic analysis of natural selection pressure in Streptococcus genomes
- PMID: 17760998
- PMCID: PMC2031904
- DOI: 10.1186/1471-2148-7-154
Phylogenomic analysis of natural selection pressure in Streptococcus genomes
Abstract
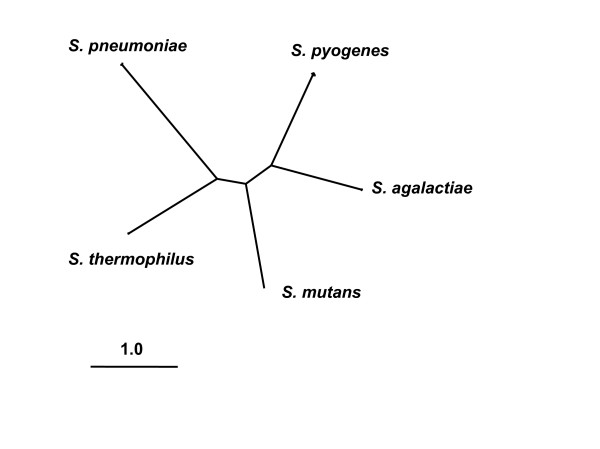
Background: In comparative analyses of bacterial pathogens, it has been common practice to discriminate between two types of genes: (i) those shared by pathogens and their non-pathogenic relatives (core genes), and (ii) those found exclusively in pathogens (pathogen-specific accessory genes). Rather than attempting to a priori delineate genes into sets more or less relevant to pathogenicity, we took a broad approach to the analysis of Streptococcus species by investigating the strength of natural selection in all clusters of homologous genes. The genus Streptococcus is comprised of a wide variety of both pathogenic and commensal lineages, and we relate our findings to the pre-existing knowledge of Streptococcus virulence factors.
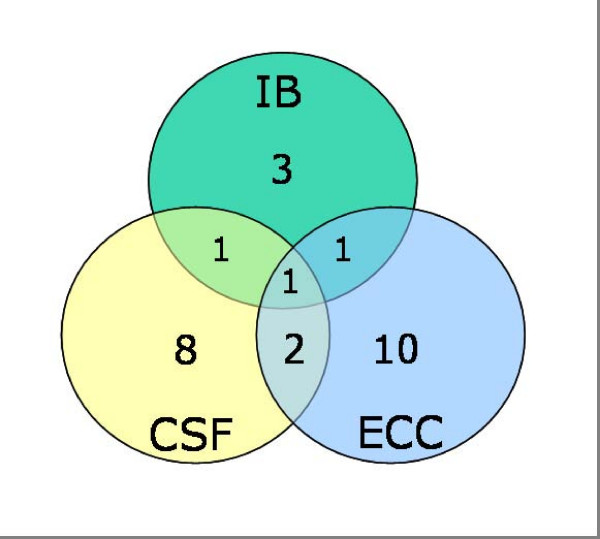
Results: Our analysis of 1730 gene clusters revealed 136 cases of positive Darwinian selection, which we suggest is most likely to result from an antagonistic interaction between the host and pathogen at the molecular level. A two-step validation procedure suggests that positive selection was robustly identified in our genomic survey. We found no evidence to support the notion that pathogen specific accessory genes are more likely to be subject to positive selection than core genes. Indeed, we even uncovered a few cases of essential gene evolution by positive selection. Among the gene clusters subject to positive selection, a large fraction (29%) can be connected to virulence. The most striking finding was that a considerable fraction of the positively selected genes are also known to have tissue specific patterns of expression during invasive disease. As current expression data is far from comprehensive, we suggest that this fraction was underestimated.
Conclusion: Our findings suggest that pathogen specific genes, although a popular focus of research, do not provide a complete picture of the evolutionary dynamics of virulence. The results of this study, and others, support the notion that the products of both core and accessory genes participate in complex networks that comprise the molecular basis of virulence. Future work should seek to understand the evolutionary dynamics of both core and accessory genes as a function of the networks in which they participate.
Figures
References
-
- Gill SR, Fouts DE, Archer GL, Mongodin EF, Deboy RT, Ravel J, Paulsen IT, Kolonay JF, Brinkac L, Beanan M, Dodson RJ, Daugherty SC, Madupu R, Angiuoli SV, Durkin AS, Haft DH, Vamathevan J, Khouri H, Utterback T, Lee C, Dimitrov G, Jiang L, Qin H, Weidman J, Tran K, Kang K, Hance IR, Nelson KE, Fraser CM. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J Bacteriol. 2005;187:2426–2438. doi: 10.1128/JB.187.7.2426-2438.2005. - DOI - PMC - PubMed
-
- Chen SL, Hung CS, Xu J, Reigstad CS, Magrini V, Sabo A, Blasiar D, Bieri T, Meyer RR, Ozersky P, Armstrong JR, Fulton RS, Latreille JP, Spieth J, Hooton TM, Mardis ER, Hultgren SJ, Gordon JI. Identification of genes subject to positive selection in uropathogenic strains of Escherichia coli: a comparative genomics approach. Proc Natl Acad Sci USA. 2006;103:5977–82. doi: 10.1073/pnas.0600938103. - DOI - PMC - PubMed
-
- Ruoff K, Whiley RA, Beighton D. Streptococcus. In: Murray PR, Baron EJ, Jorgensen JH, Pfaller MA, Yolken RH, editor. Manual of clinical microbiology. 8. Vol. 1. Washington, D.C: ASM Press; 2003. pp. 405–421.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
