

GeneSrF and varSelRF: a web-based tool and R package for gene selection and classification using random forest
- PMID: 17767709
- PMCID: PMC2034606
- DOI: 10.1186/1471-2105-8-328
GeneSrF and varSelRF: a web-based tool and R package for gene selection and classification using random forest
Abstract
Background: Microarray data are often used for patient classification and gene selection. An appropriate tool for end users and biomedical researchers should combine user friendliness with statistical rigor, including carefully avoiding selection biases and allowing analysis of multiple solutions, together with access to additional functional information of selected genes. Methodologically, such a tool would be of greater use if it incorporates state-of-the-art computational approaches and makes source code available.
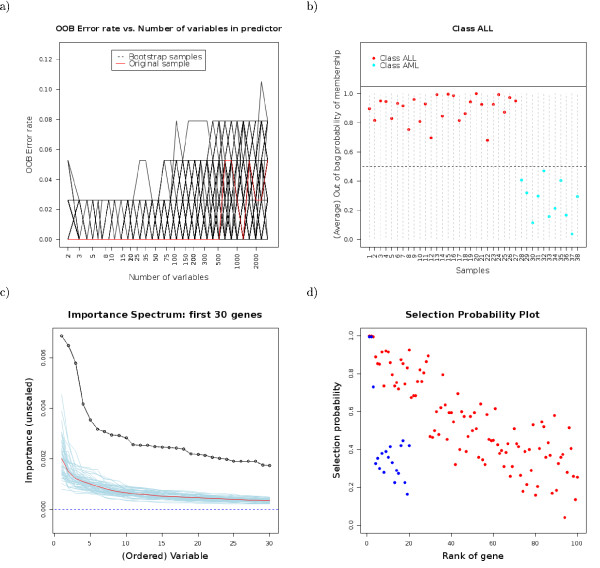
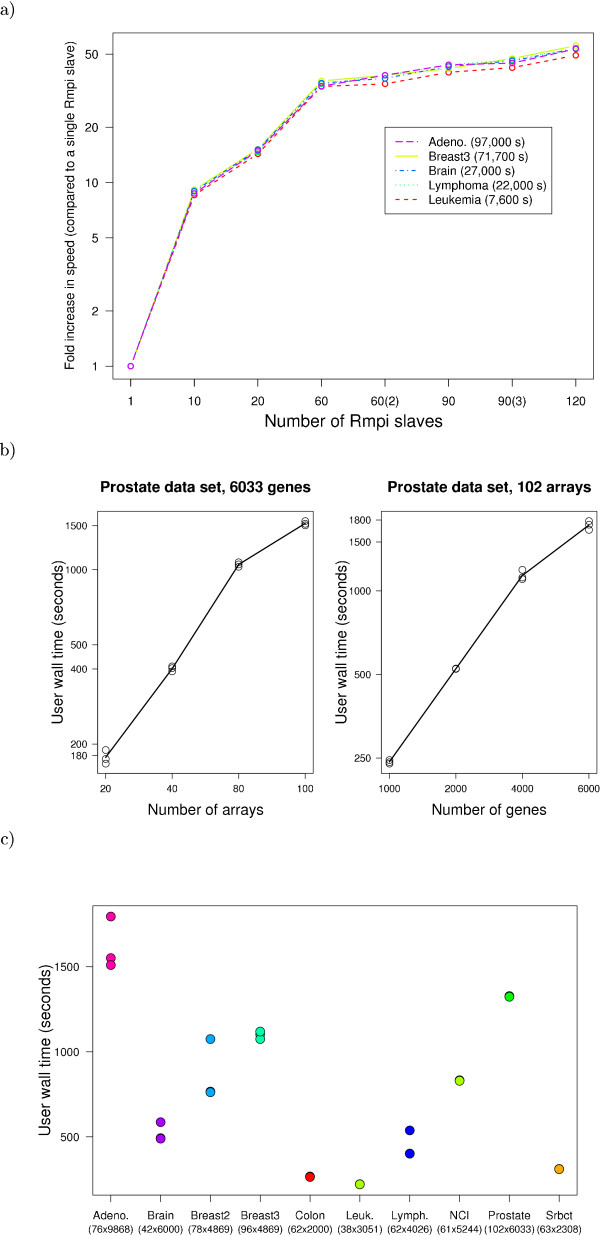
Results: We have developed GeneSrF, a web-based tool, and varSelRF, an R package, that implement, in the context of patient classification, a validated method for selecting very small sets of genes while preserving classification accuracy. Computation is parallelized, allowing to take advantage of multicore CPUs and clusters of workstations. Output includes bootstrapped estimates of prediction error rate, and assessments of the stability of the solutions. Clickable tables link to additional information for each gene (GO terms, PubMed citations, KEGG pathways), and output can be sent to PaLS for examination of PubMed references, GO terms, KEGG and and Reactome pathways characteristic of sets of genes selected for class prediction. The full source code is available, allowing to extend the software. The web-based application is available from http://genesrf2.bioinfo.cnio.es. All source code is available from Bioinformatics.org or The Launchpad. The R package is also available from CRAN.
Conclusion: varSelRF and GeneSrF implement a validated method for gene selection including bootstrap estimates of classification error rate. They are valuable tools for applied biomedical researchers, specially for exploratory work with microarray data. Because of the underlying technology used (combination of parallelization with web-based application) they are also of methodological interest to bioinformaticians and biostatisticians.
Figures
References
-
- Simon R, Radmacher MD, Dobbin K, McShane LM. Pitfalls in the use of DNA microarray data for diagnostic and prognostic classification. Journal of the National Cancer Institute. 2003;95:14–18. - PubMed
-
- Dudoit S, Fridlyand J. Classification in microarray experiments. In: Speed T, editor. Statistical analysis of gene expression microarray data. New York: Chapman & Hall; 2003. pp. 93–158.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
