

New analysis for consistency among markers in the study of genetic diversity: development and application to the description of bacterial diversity
- PMID: 17767711
- PMCID: PMC2082430
- DOI: 10.1186/1471-2148-7-156
New analysis for consistency among markers in the study of genetic diversity: development and application to the description of bacterial diversity
Abstract
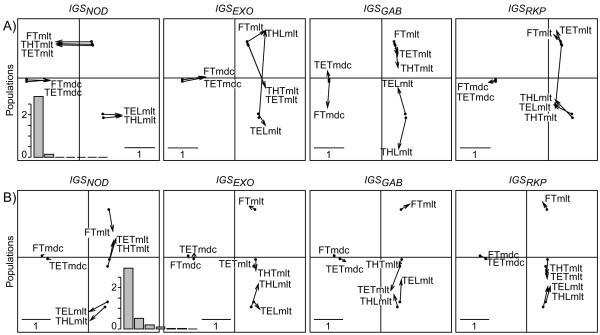
Background: The development of post-genomic methods has dramatically increased the amount of qualitative and quantitative data available to understand how ecological complexity is shaped. Yet, new statistical tools are needed to use these data efficiently. In support of sequence analysis, diversity indices were developed to take into account both the relative frequencies of alleles and their genetic divergence. Furthermore, a method for describing inter-population nucleotide diversity has recently been proposed and named the double principal coordinate analysis (DPCoA), but this procedure can only be used with one locus. In order to tackle the problem of measuring and describing nucleotide diversity with more than one locus, we developed three versions of multiple DPCoA by using three ordination methods: multiple co-inertia analysis, STATIS, and multiple factorial analysis.
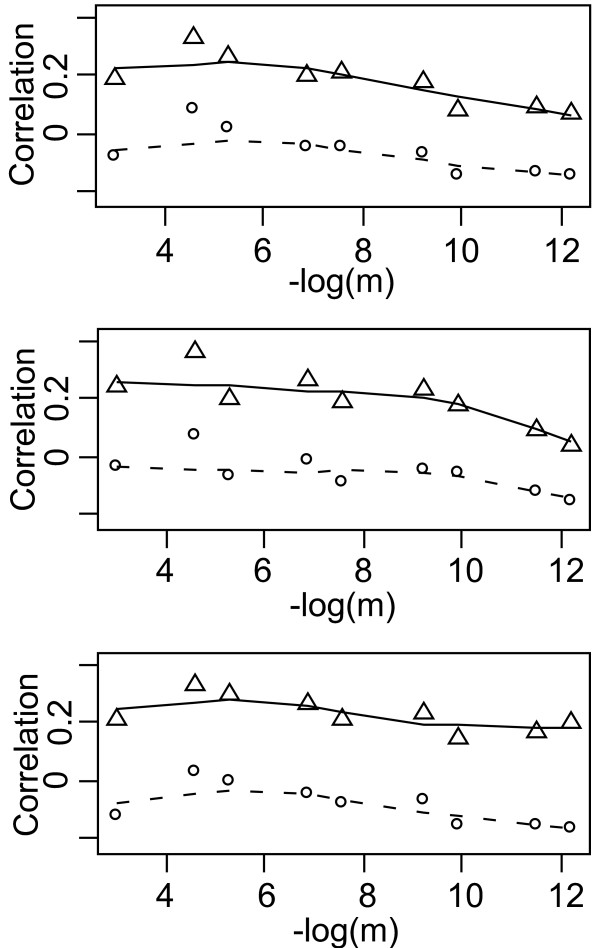
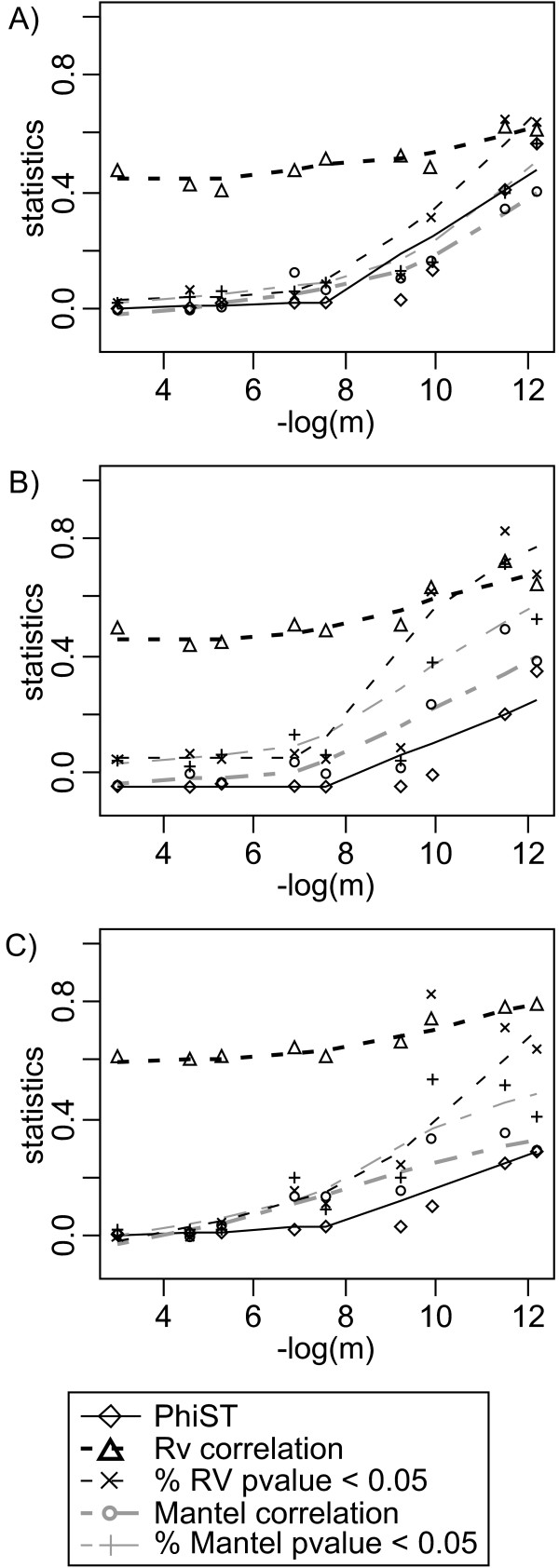
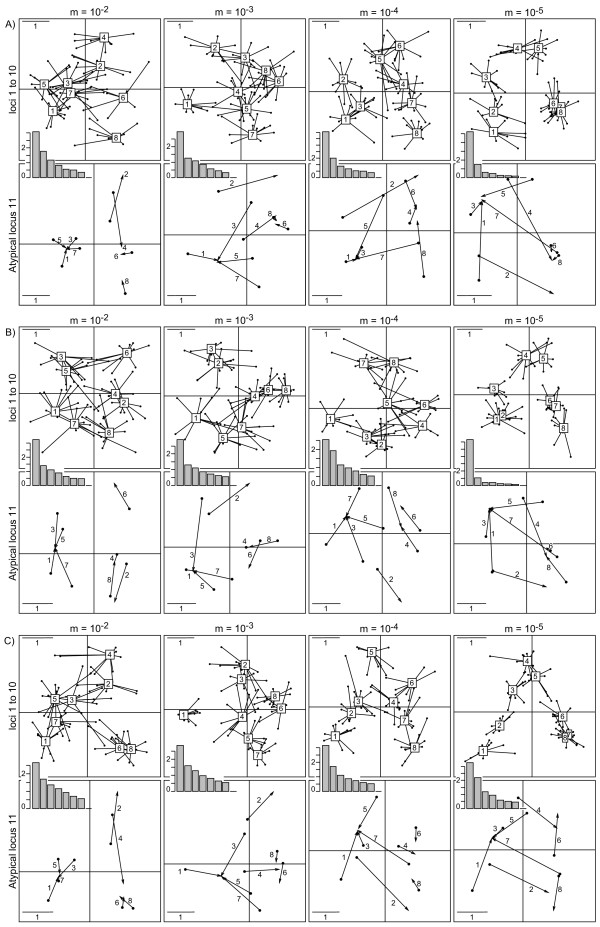
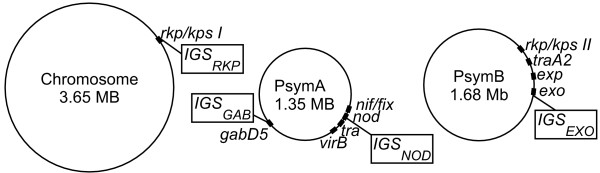
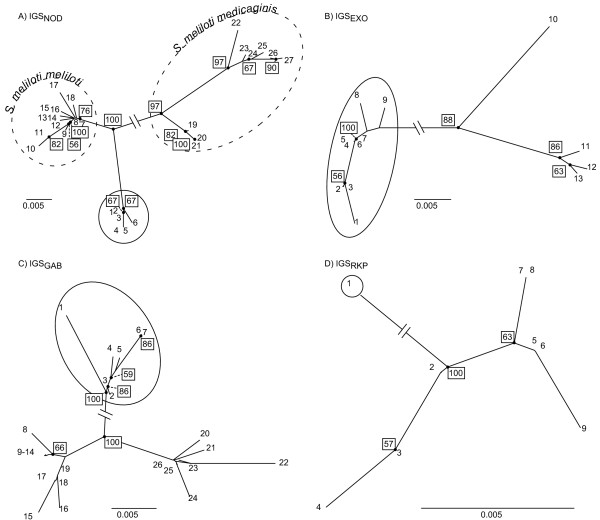
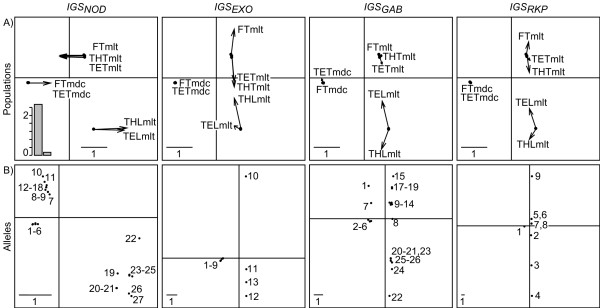
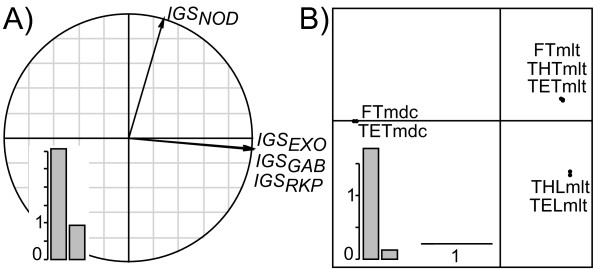
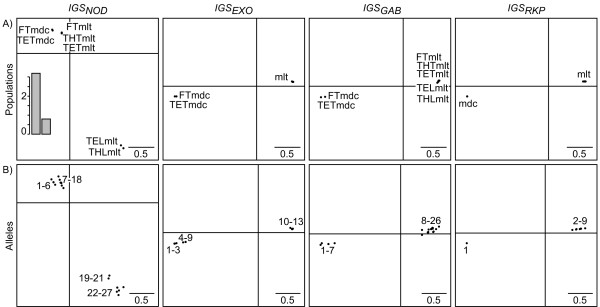
Results: This combination of methods allows i) testing and describing differences in patterns of inter-population diversity among loci, and ii) defining the best compromise among loci. These methods are illustrated by the analysis of both simulated data sets, which include ten loci evolving under a stepping stone model and a locus evolving under an alternative population structure, and a real data set focusing on the genetic structure of two nitrogen fixing bacteria, which is influenced by geographical isolation and host specialization. All programs needed to perform multiple DPCoA are freely available.
Conclusion: Multiple DPCoA allows the evaluation of the impact of various loci in the measurement and description of diversity. This method is general enough to handle a large variety of data sets. It complements existing methods such as the analysis of molecular variance or other analyses based on linkage disequilibrium measures, and is very useful to study the impact of various loci on the measurement of diversity.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
