

Tension-sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore microtubule interactions
- PMID: 17785528
- PMCID: PMC1950859
- DOI: 10.1101/gad.436007
Tension-sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore microtubule interactions
Abstract
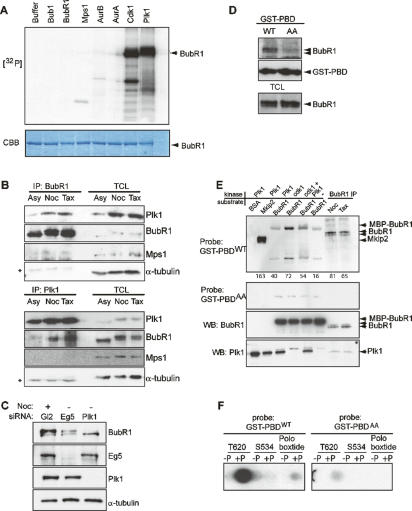
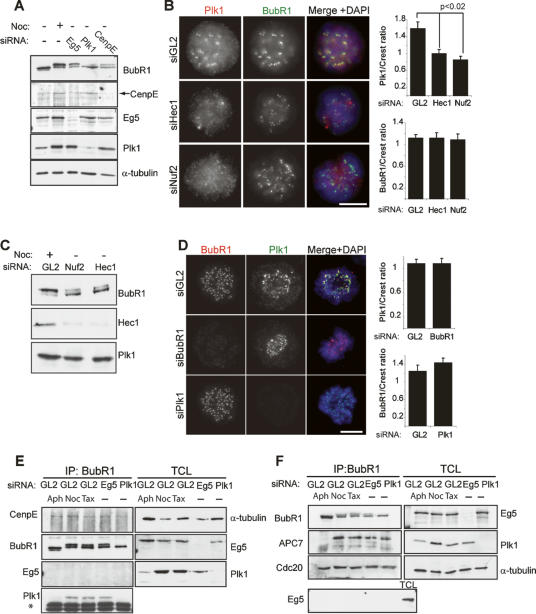
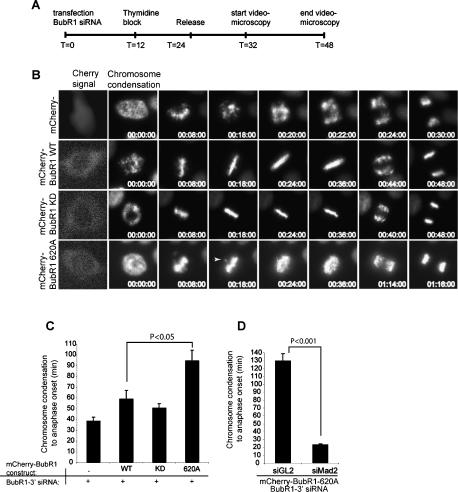
Mitotic phosphorylation of the spindle checkpoint component BubR1 is highly conserved throughout evolution. Here, we demonstrate that BubR1 is phosphorylated on the Cdk1 site T620, which triggers the recruitment of Plk1 and phosphorylation of BubR1 by Plk1 both in vitro and in vivo. Phosphorylation does not appear to be required for spindle checkpoint function but instead is important for the stability of kinetochore-microtubule (KT-MT) interactions, timely mitotic progression, and chromosome alignment onto the metaphase plate. By quantitative mass spectrometry, we identify S676 as a Plk1-specific phosphorylation site on BubR1. Furthermore, using a phospho-specific antibody, we show that this site is phosphorylated during prometaphase, but dephosphorylated at metaphase upon establishment of tension between sister chromatids. These findings describe the first in vivo verified phosphorylation site for human BubR1, identify Plk1 as the kinase responsible for causing the characteristic mitotic BubR1 upshift, and attribute a KT-specific function to the hyperphosphorylated form of BubR1 in the stabilization of KT-MT interactions.
Figures




References
-
- Abrieu A., Kahana J.A., Wood K.W., Cleveland D.W., Kahana J.A., Wood K.W., Cleveland D.W., Wood K.W., Cleveland D.W., Cleveland D.W. CENP-E as an essential component of the mitotic checkpoint in vitro. Cell. 2000;102:817–826. - PubMed
-
- Ahonen L.J., Kallio M.J., Daum J.R., Bolton M., Manke I.A., Yaffe M.B., Stukenberg P.T., Gorbsky G.J., Kallio M.J., Daum J.R., Bolton M., Manke I.A., Yaffe M.B., Stukenberg P.T., Gorbsky G.J., Daum J.R., Bolton M., Manke I.A., Yaffe M.B., Stukenberg P.T., Gorbsky G.J., Bolton M., Manke I.A., Yaffe M.B., Stukenberg P.T., Gorbsky G.J., Manke I.A., Yaffe M.B., Stukenberg P.T., Gorbsky G.J., Yaffe M.B., Stukenberg P.T., Gorbsky G.J., Stukenberg P.T., Gorbsky G.J., Gorbsky G.J. Polo-like kinase 1 creates the tension-sensing 3F3/2 phosphoepitope and modulates the association of spindle-checkpoint proteins at kinetochores. Curr. Biol. 2005;15:1078–1089. - PubMed
-
- Barr F.A., Sillje H.H., Nigg E.A., Sillje H.H., Nigg E.A., Nigg E.A. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 2004;5:429–440. - PubMed
-
- Baumann C., Korner R., Hofmann K., Nigg E.A., Korner R., Hofmann K., Nigg E.A., Hofmann K., Nigg E.A., Nigg E.A. PICH, a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell. 2007;128:101–114. - PubMed
-
- Bharadwaj R., Yu H., Yu H. The spindle checkpoint, aneuploidy, and cancer. Oncogene. 2004;23:2016–2027. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous