

The evolution of genome compression and genomic novelty in RNA viruses
- PMID: 17785537
- PMCID: PMC1987338
- DOI: 10.1101/gr.6305707
The evolution of genome compression and genomic novelty in RNA viruses
Abstract
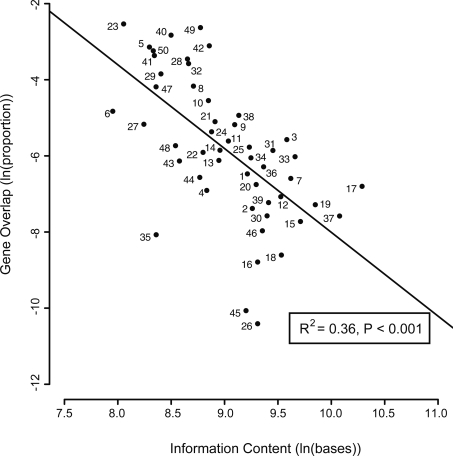
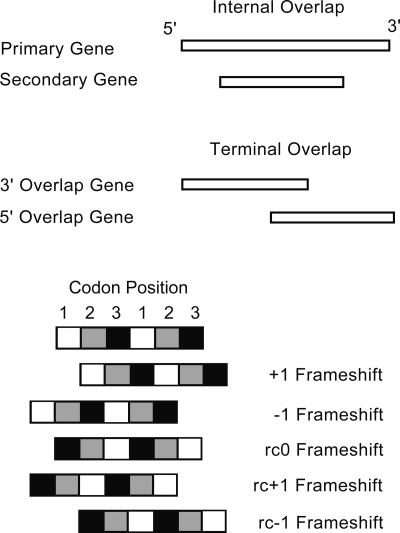
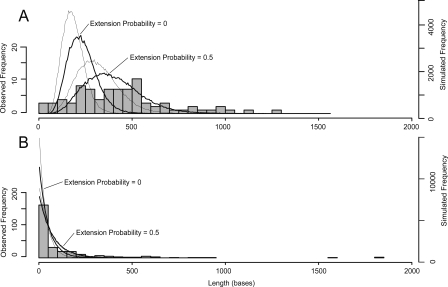
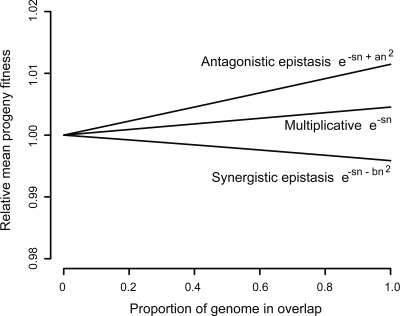
The genomes of RNA viruses are characterized by their extremely small size and extremely high mutation rates (typically 10 kb and 10(-4)/base/replication cycle, respectively), traits that are thought to be causally linked. One aspect of their small size is the genome compression caused by the use of overlapping genes (where some nucleotides code for two genes). Using a comparative analysis of all known RNA viral species, we show that viruses with larger genomes tend to have less gene overlap. We provide a numerical model to show how a high mutation rate could lead to gene overlap, and we discuss the factors that might explain the observed relationship between gene overlap and genome size. We also propose a model for the evolution of gene overlap based on the co-opting of previously unused ORFs, which gives rise to two types of overlap: (1) the creation of novel genes inside older genes, predominantly via +1 frameshifts, and (2) the incremental increase in overlap between originally contiguous genes, with no frameshift preference. Both types of overlap are viewed as the creation of genomic novelty under pressure for genome compression. Simulations based on our model generate the empirical size distributions of overlaps and explain the observed frameshift preferences. We suggest that RNA viruses are a good model system for the investigation of general evolutionary relationship between genome attributes such as mutational robustness, mutation rate, and size.
Figures
References
-
- Burch C.L., Turner P.E., Hanley K.A., Turner P.E., Hanley K.A., Hanley K.A. Patterns of epistasis in RNA viruses: A review of the evidence from vaccine design. J. Evol. Biol. 2003;16:1223–1235. - PubMed
-
- Chare E.R., Gould E.A., Holmes E.C., Gould E.A., Holmes E.C., Holmes E.C. Phylogenetic analysis reveals a low rate of homologous recombination in negative-sense RNA viruses. J. Gen. Virol. 2003;84:2691–2703. - PubMed
-
- Codoñer F.M., Daròs J.A., Solé R.V., Elena S.F., Daròs J.A., Solé R.V., Elena S.F., Solé R.V., Elena S.F., Elena S.F. The fittest versus the flattest: Experimental confirmation of the quasispecies effect with subviral pathogens. PLoS Pathog. 2006;2:e136. doi: 10.1371/journal.ppat.0020136. - DOI - PMC - PubMed
-
- Cristina J., Lopez F., Moratorio G., Lopez L., Vasquez S., Garcia-Aguirre L., Chunga A., Lopez F., Moratorio G., Lopez L., Vasquez S., Garcia-Aguirre L., Chunga A., Moratorio G., Lopez L., Vasquez S., Garcia-Aguirre L., Chunga A., Lopez L., Vasquez S., Garcia-Aguirre L., Chunga A., Vasquez S., Garcia-Aguirre L., Chunga A., Garcia-Aguirre L., Chunga A., Chunga A. Hepatitis C virus F protein sequence reveals a lack of functional constraints and a variable pattern of amino acid substitution. J. Gen. Virol. 2005;86:115–120. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources