

The diploid genome sequence of an individual human
- PMID: 17803354
- PMCID: PMC1964779
- DOI: 10.1371/journal.pbio.0050254
The diploid genome sequence of an individual human
Abstract
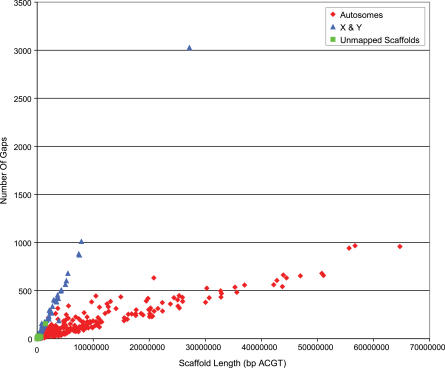
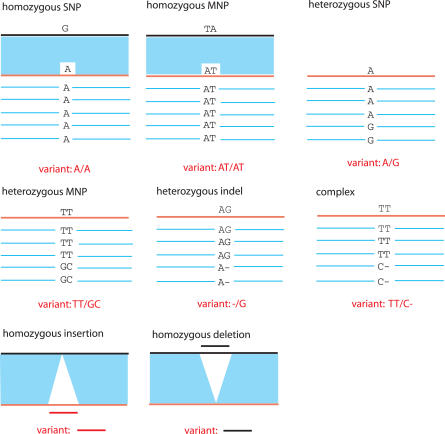
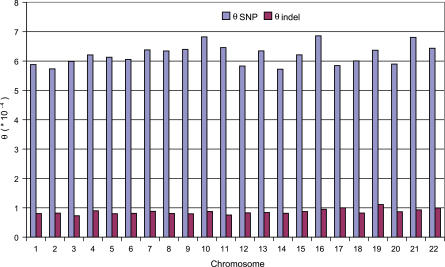
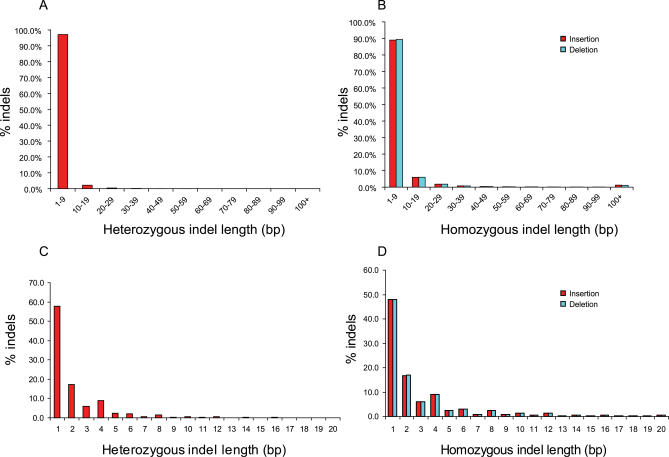
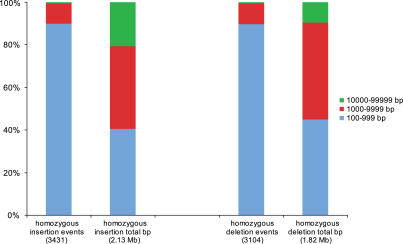
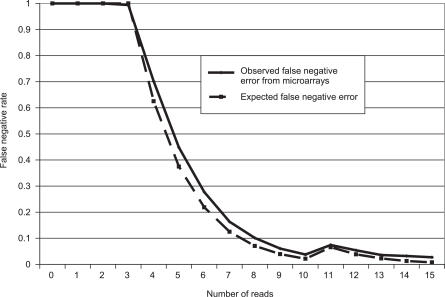
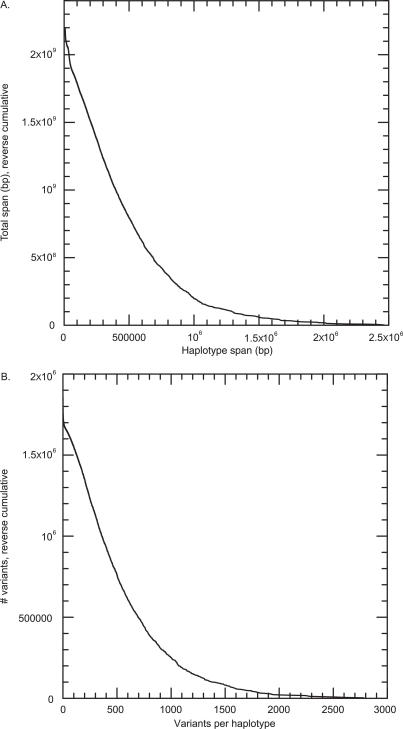
Presented here is a genome sequence of an individual human. It was produced from approximately 32 million random DNA fragments, sequenced by Sanger dideoxy technology and assembled into 4,528 scaffolds, comprising 2,810 million bases (Mb) of contiguous sequence with approximately 7.5-fold coverage for any given region. We developed a modified version of the Celera assembler to facilitate the identification and comparison of alternate alleles within this individual diploid genome. Comparison of this genome and the National Center for Biotechnology Information human reference assembly revealed more than 4.1 million DNA variants, encompassing 12.3 Mb. These variants (of which 1,288,319 were novel) included 3,213,401 single nucleotide polymorphisms (SNPs), 53,823 block substitutions (2-206 bp), 292,102 heterozygous insertion/deletion events (indels)(1-571 bp), 559,473 homozygous indels (1-82,711 bp), 90 inversions, as well as numerous segmental duplications and copy number variation regions. Non-SNP DNA variation accounts for 22% of all events identified in the donor, however they involve 74% of all variant bases. This suggests an important role for non-SNP genetic alterations in defining the diploid genome structure. Moreover, 44% of genes were heterozygous for one or more variants. Using a novel haplotype assembly strategy, we were able to span 1.5 Gb of genome sequence in segments >200 kb, providing further precision to the diploid nature of the genome. These data depict a definitive molecular portrait of a diploid human genome that provides a starting point for future genome comparisons and enables an era of individualized genomic information.
Conflict of interest statement
Figures
Comment in
-
A new human genome sequence paves the way for individualized genomics.PLoS Biol. 2007 Oct;5(10):e266. doi: 10.1371/journal.pbio.0050266. Epub 2007 Sep 4. PLoS Biol. 2007. PMID: 20076646 Free PMC article. No abstract available.
References
-
- Painter TS. The sex chromosomes of man. Am Nat. 1924;58:506–524.
-
- Tjio TH, Levan A. The chromosome number of man. Hereditas. 1956;42:1.
-
- Caspersson T, Zech L, Johansson C, Modest EJ. Identification of human chromosomes by DNA-binding fluorescent agents. Chromosoma. 1970;30:215–227. - PubMed
-
- Fodor SP, Read JL, Pirrung MC, Stryer L, Lu AT, et al. Light-directed, spatially addressable parallel chemical synthesis. Science. 1991;251:767–773. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
