

Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool
- PMID: 17804638
- PMCID: PMC6672978
- DOI: 10.1523/JNEUROSCI.5586-06.2007
Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool
Abstract
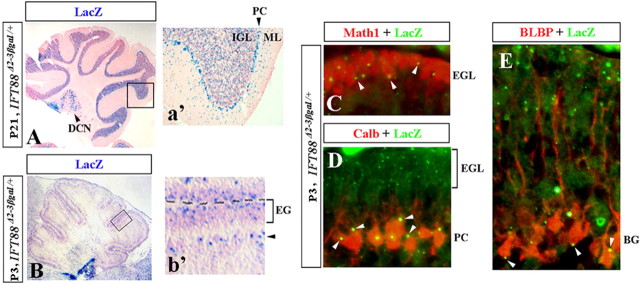
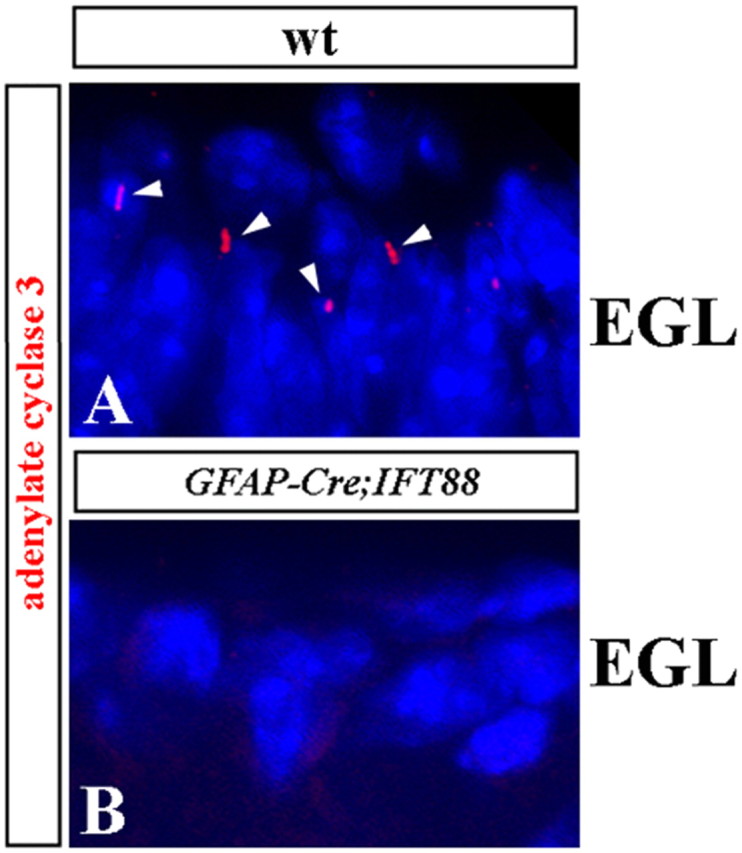
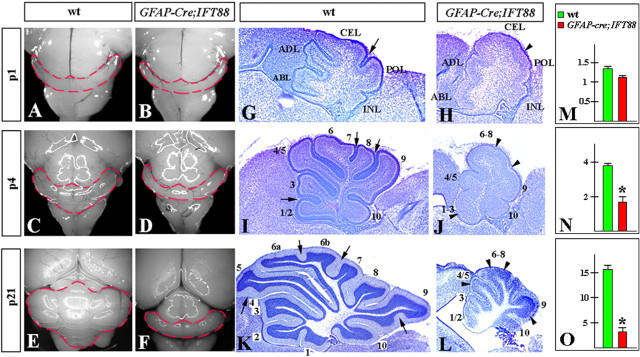
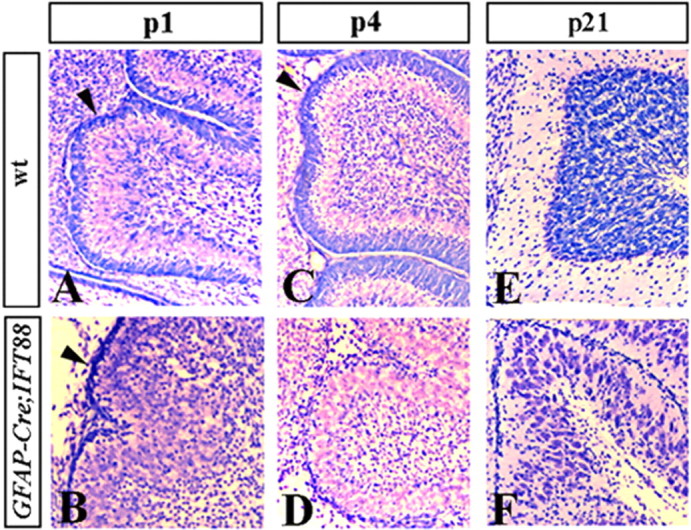
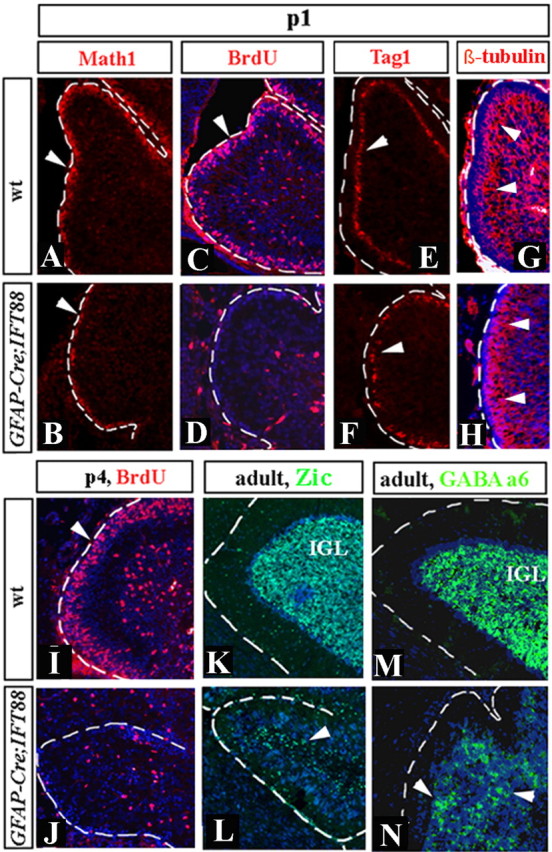
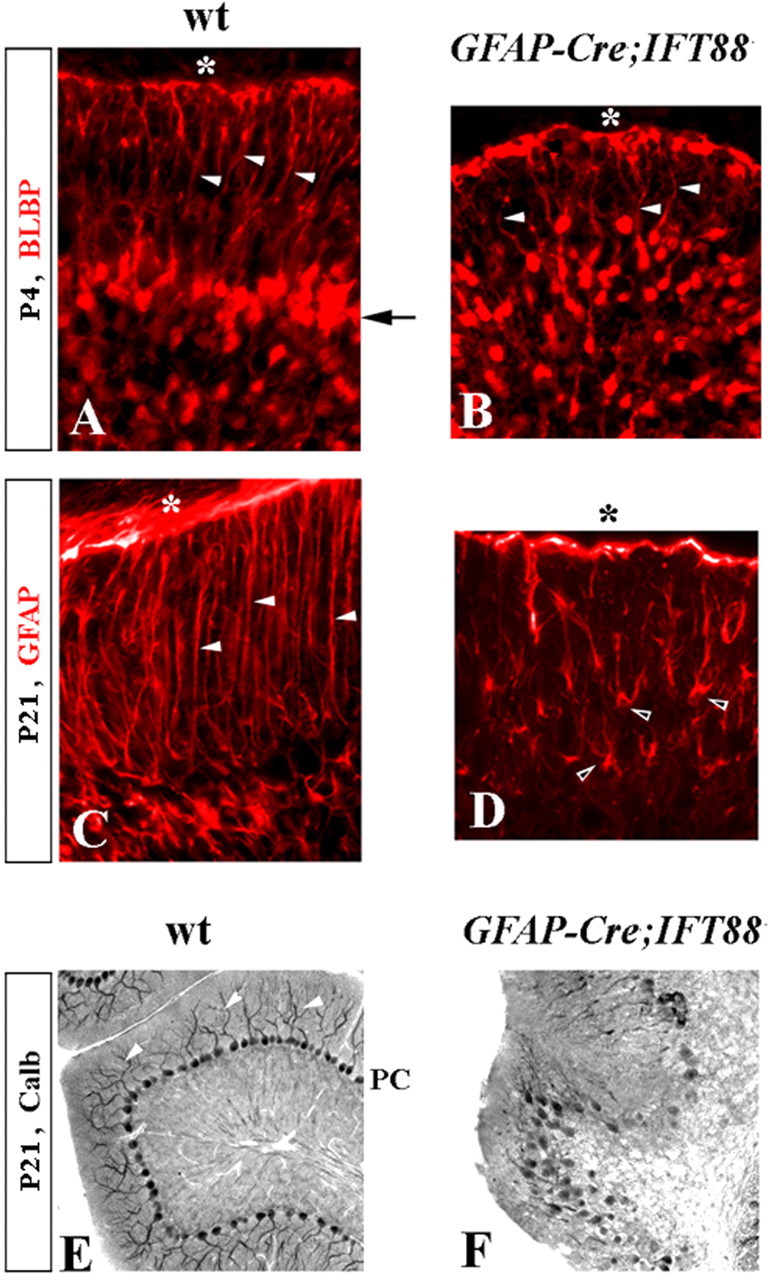
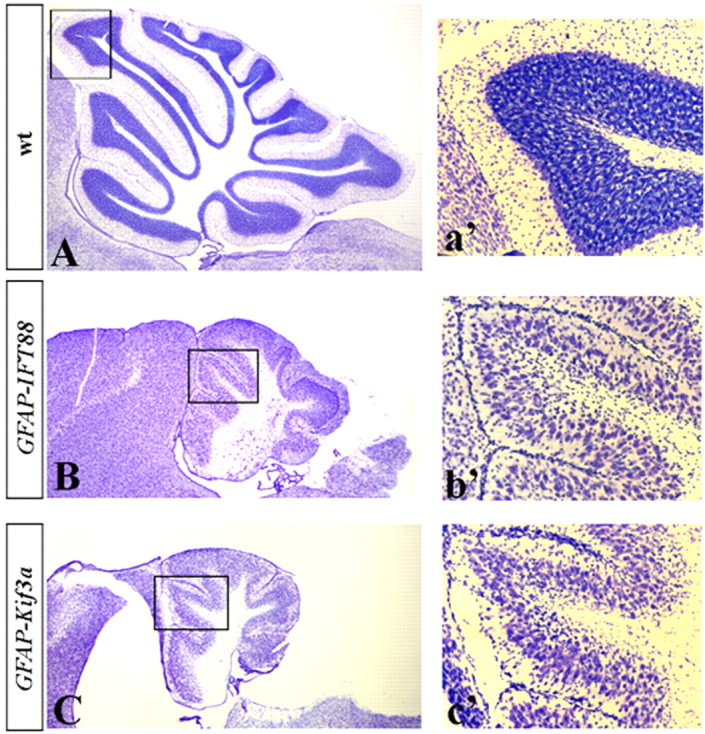
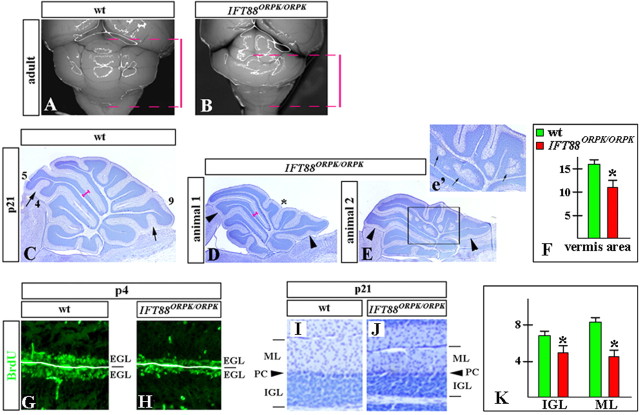
Although human congenital cerebellar malformations are common, their molecular and developmental basis is still poorly understood. Recently, cilia-related gene deficiencies have been implicated in several congenital disorders that exhibit cerebellar abnormalities such as Joubert syndrome, Meckel-Gruber syndrome, Bardet-Biedl syndrome, and Orofaciodigital syndrome. The association of cilia gene mutations with these syndromes suggests that cilia may be important for cerebellar development, but the nature of cilia involvement has not been elucidated. To assess the importance of cilia-related proteins during cerebellar development, we studied the effects of CNS-specific inactivation of two mouse genes whose protein products are critical for cilia formation and maintenance, IFT88, (also known as polaris or Tg737), which encodes intraflagellar transport 88 homolog, and Kif3a, which encodes kinesin family member 3a. We showed that loss of either of these genes caused severe cerebellar hypoplasia and foliation abnormalities, primarily attributable to a failure of expansion of the neonatal granule cell progenitor population. In addition, granule cell progenitor proliferation was sensitive to partial loss of IFT function in a hypomorphic mutant of IFT88 (IFT88(orpk)), an effect that was modified by genetic background. IFT88 and Kif3a were not required for the specification and differentiation of most other cerebellar cell types, including Purkinje cells. Together, our observations constitute the first demonstration that cilia proteins are essential for normal cerebellar development and suggest that granule cell proliferation defects may be central to the cerebellar pathology in human cilia-related disorders.
Figures
References
-
- Altman J, Bayer SA. New York: CRC; 1997. The development of the cerebellar system: in relation in its evolution, structure and function.
-
- Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Leitch CC, Kim JC, Ross AJ, Eichers ER, Teslovich TM, Mah AK, Johnsen RC, Cavender JC, Lewis RA, Leroux MR, Beales PL, Katsanis N. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature. 2003;425:628–633. - PubMed
-
- Arts HH, Doherty D, van Beersum SE, Parisi MA, Letteboer SJ, Gorden NT, Peters TA, Marker T, Voesenek K, Kartono A, Ozyurek H, Farin FM, Kroes HY, Wolfrum U, Brunner HG, Cremers FP, Glass IA, Knoers NV, Roepman R. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet. 2007;39:882–888. - PubMed
-
- Baala L, Romano S, Khaddour R, Saunier S, Smith UM, Audollent S, Ozilou C, Faivre L, Laurent N, Foliguet B, Munnich A, Lyonnet S, Salomon R, Encha-Razavi F, Gubler M-C, Boddaert N, de Lonlay P, Johnson CA, Vekemans M, Antignac C, Attié-Bitach T. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet. 2007;80:186–194. - PMC - PubMed
-
- Baptista CA, Hatten ME, Blazeski R, Mason CA. Cell-cell interactions influence survival and differentiation of purified Purkinje cells in vitro. Neuron. 1994;12:243–260. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases