

Late sodium current in failing heart: friend or foe?
- PMID: 17854868
- PMCID: PMC2267741
- DOI: 10.1016/j.pbiomolbio.2007.07.010
Late sodium current in failing heart: friend or foe?
Abstract
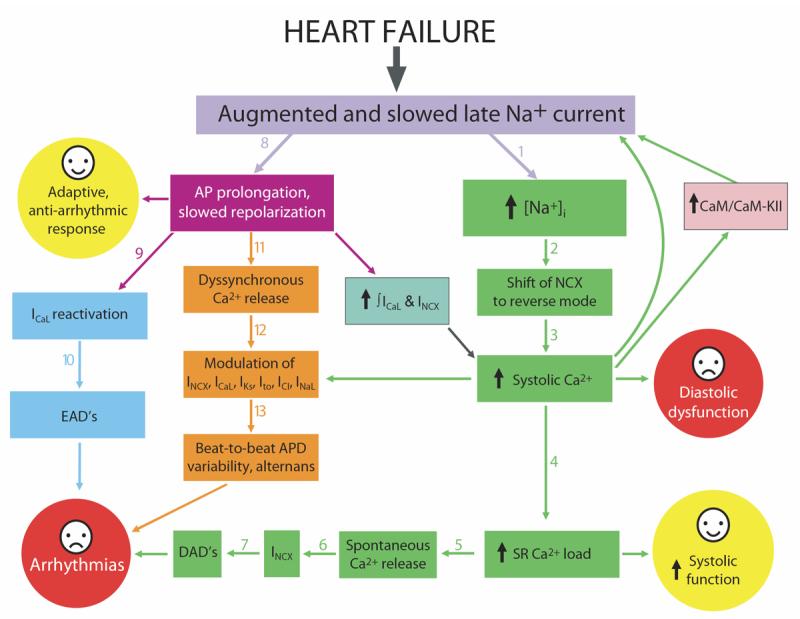
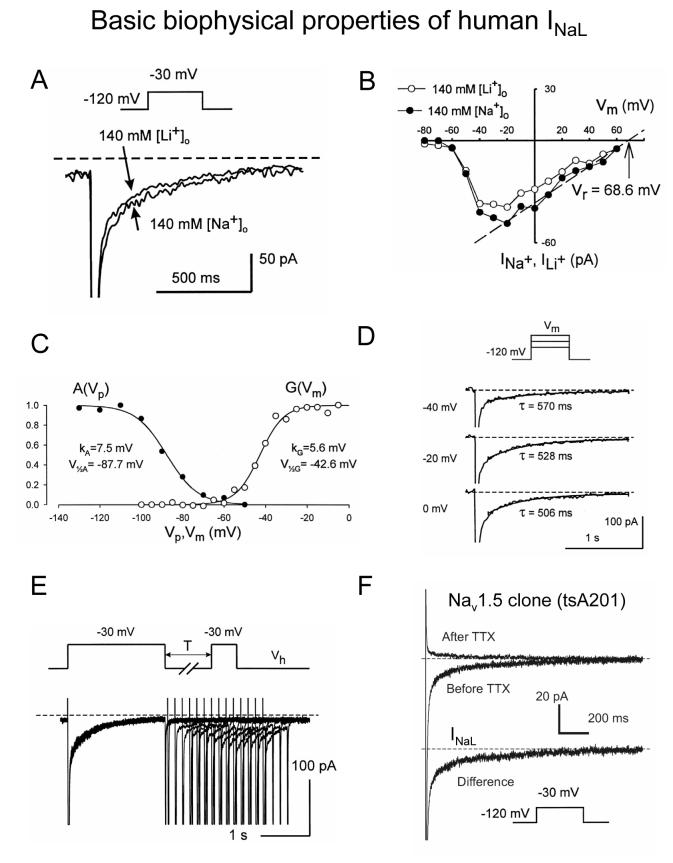
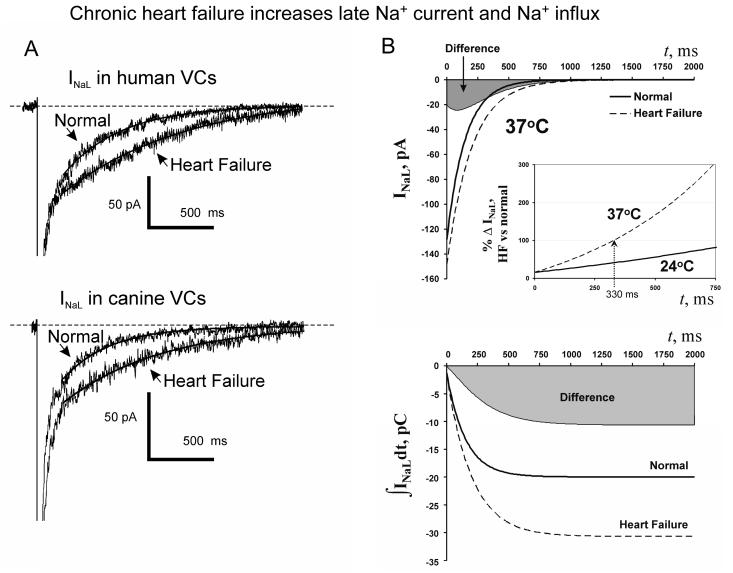
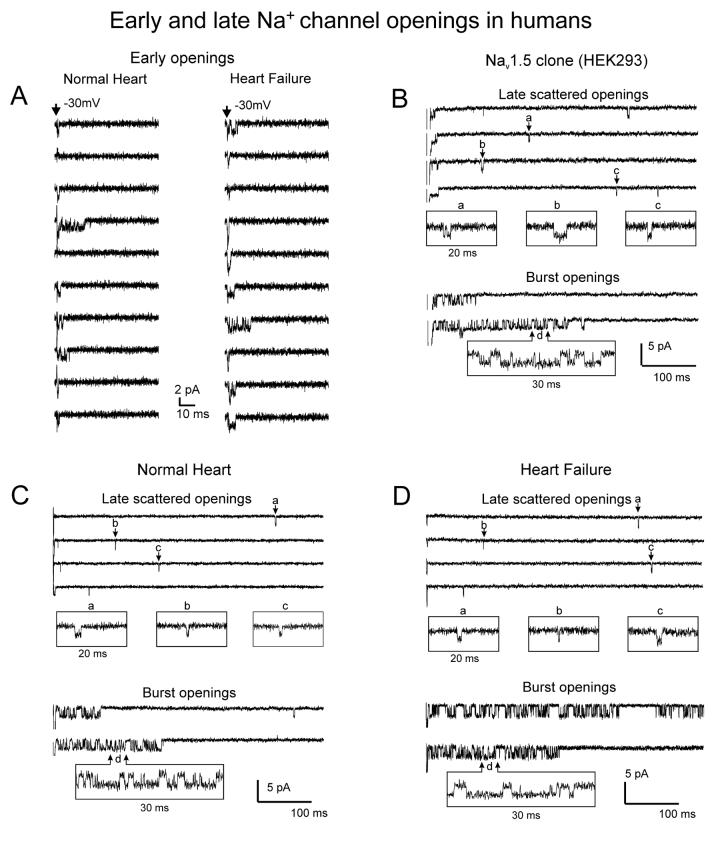
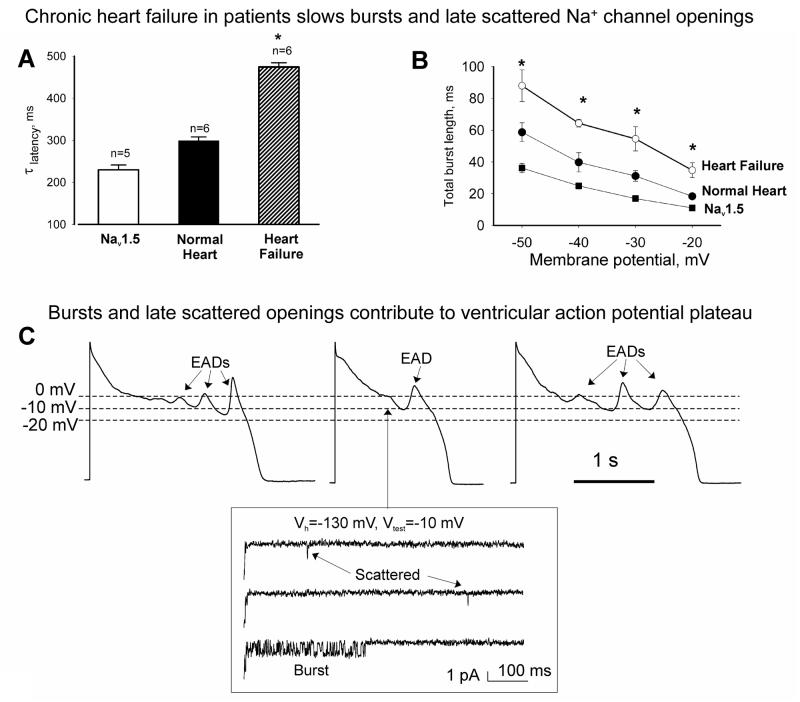
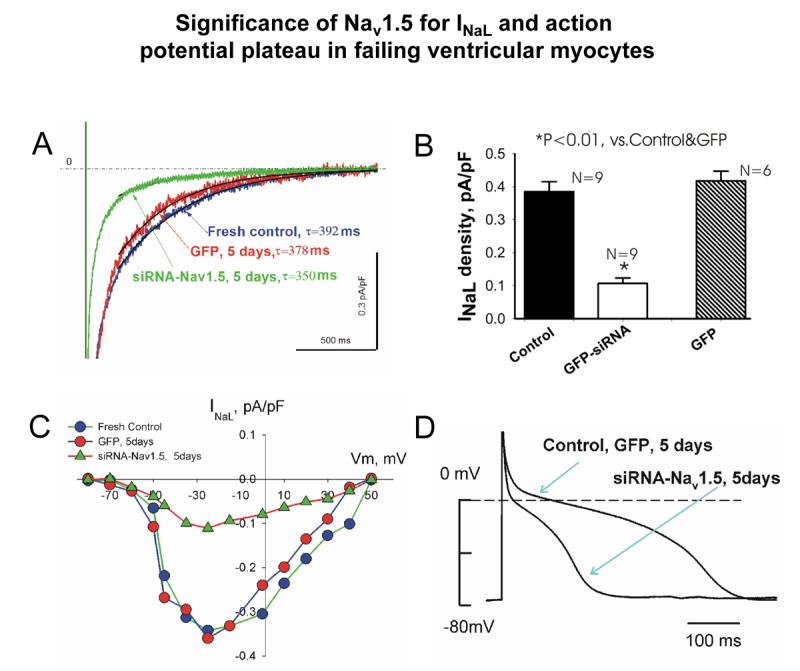
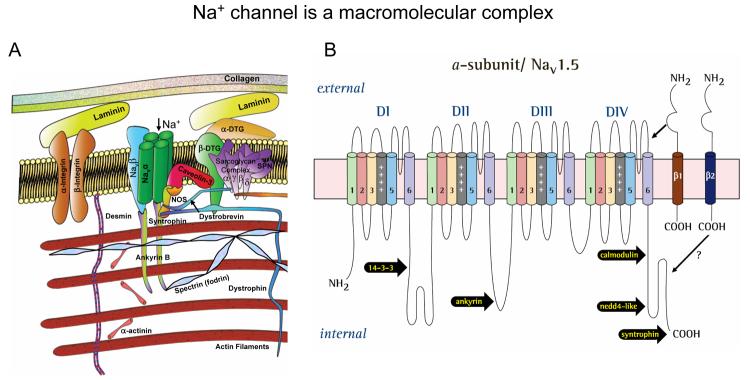
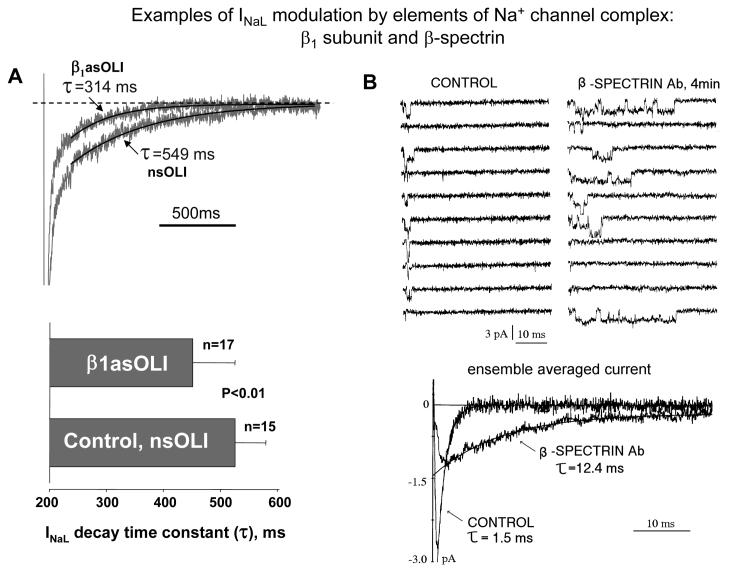
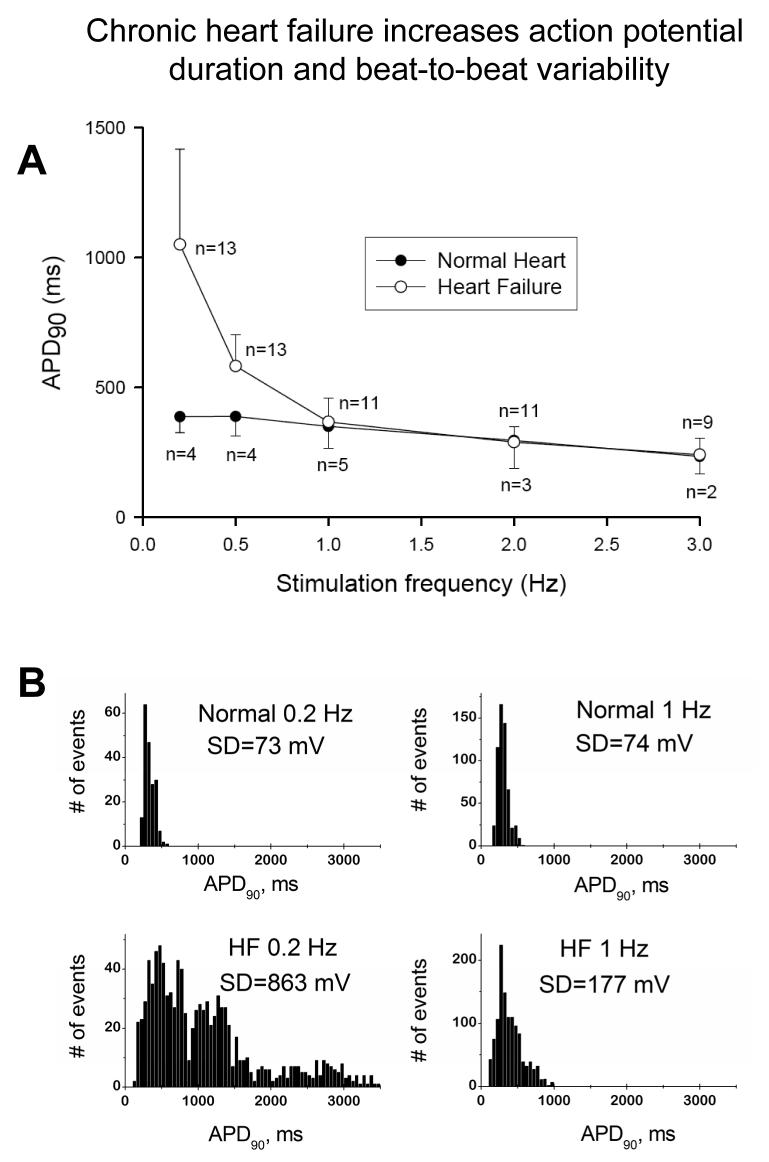
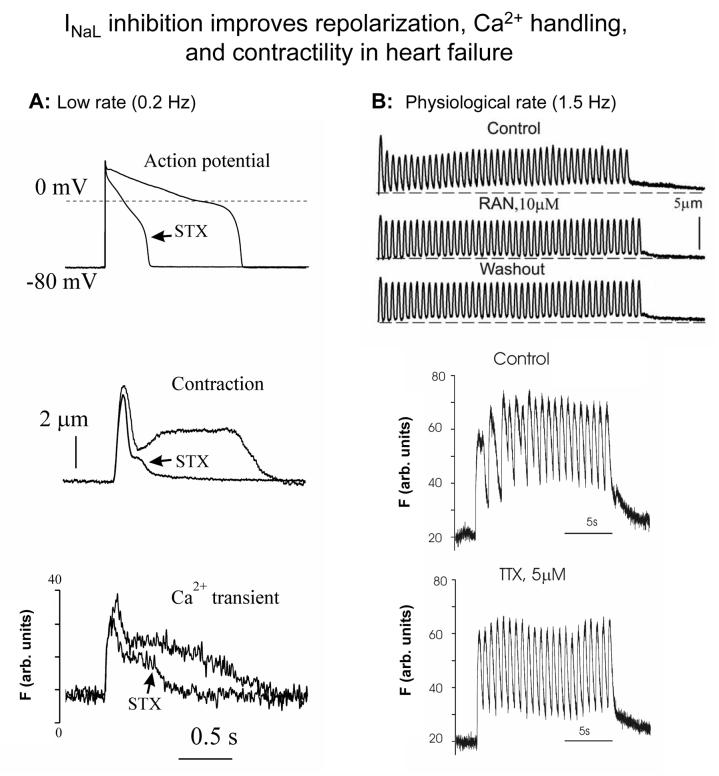
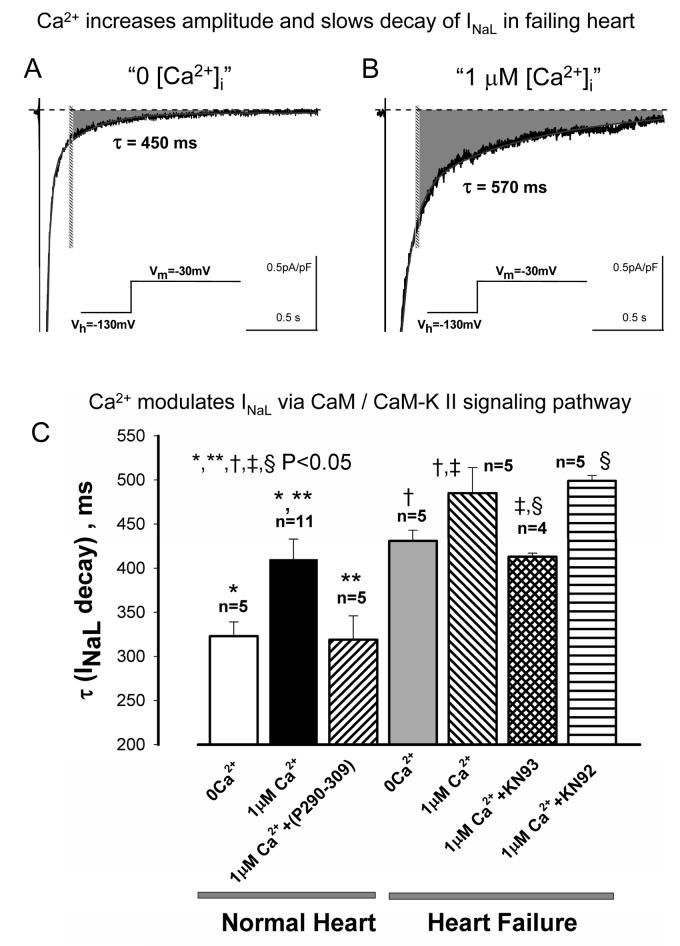
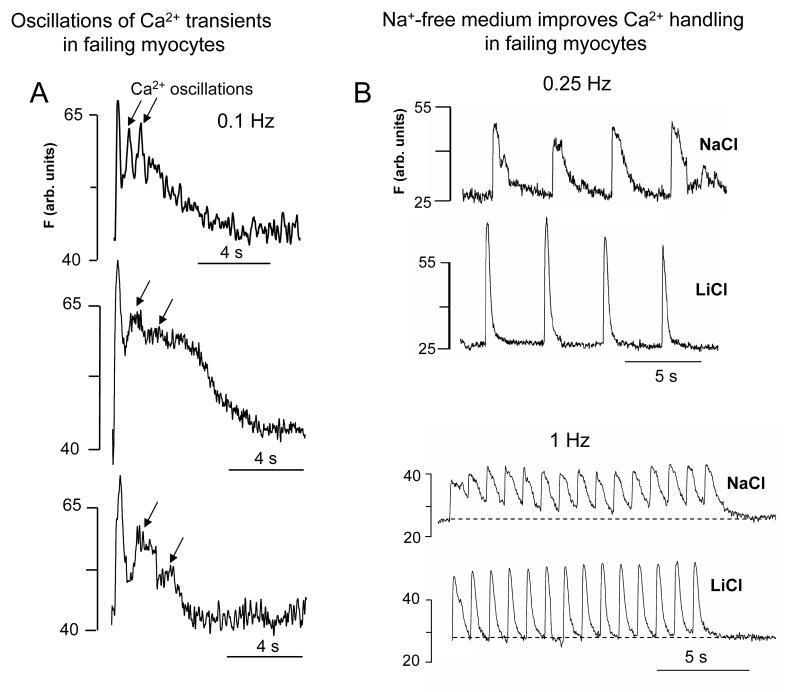
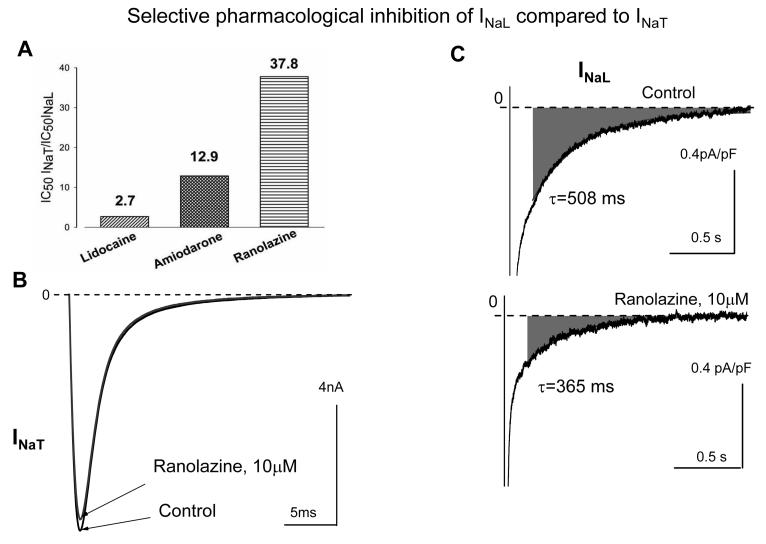
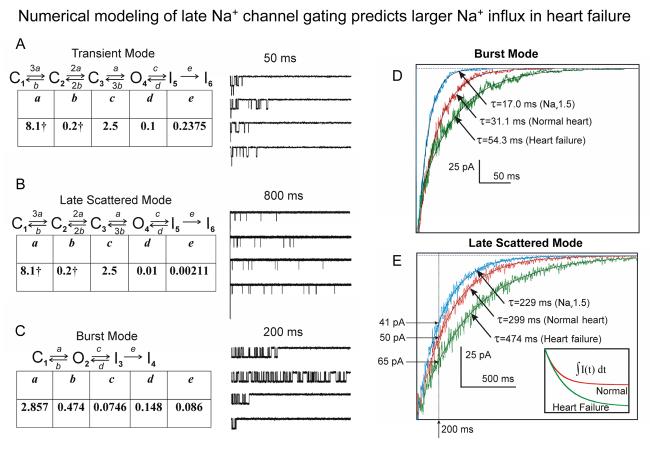
Most cardiac Na+ channels open transiently upon membrane depolarization and then are quickly inactivated. However, some channels remain active, carrying the so-called persistent or late Na+ current (INaL) throughout the action potential (AP) plateau. Experimental data and the results of numerical modeling accumulated over the past decade show the emerging importance of this late current component for the function of both normal and failing myocardium. INaL is produced by special gating modes of the cardiac-specific Na+ channel isoform. Heart failure (HF) slows channel gating and increases INaL, but HF-specific Na+ channel isoform underlying these changes has not been found. Na+ channels represent a multi-protein complex and its activity is determined not only by the pore-forming alpha subunit but also by its auxiliary beta subunits, cytoskeleton, calmodulin, regulatory kinases and phosphatases, and trafficking proteins. Disruption of the integrity of this protein complex may lead to alterations of INaL in pathological conditions. Increased INaL and the corresponding Na+ flux in failing myocardium contribute to abnormal repolarization and an increased cell Ca2+ load. Interventions designed to correct INaL rescue normal repolarization and improve Ca2+ handling and contractility of the failing cardiomyocytes. This review considers (1) quantitative integration of INaL into the established electrophysiological and Ca2+ regulatory mechanisms in normal and failing cardiomyocytes and (2) a new therapeutic strategy utilizing a selective inhibition of INaL to target both arrhythmias and impaired contractility in HF.
Figures
References
-
- Abriel H, Kass RS. Regulation of the voltage-gated cardiac sodium channel Nav1.5 by interacting proteins. Trends Cardiovasc Med. 2005;15:35–40. - PubMed
-
- Ahern CA, Zhang JF, Wookalis MJ, Horn R. Modulation of the cardiac sodium channel NaV1.5 by Fyn, a Src family tyrosine kinase. Circ Res. 2005;96:991–998. Epub 2005 Apr 2014. - PubMed
-
- Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressedy sensory neurones. Nature. 1996;379:257–262. - PubMed
-
- Allouis M, Le Bouffant F, Wilders R, Peroz D, Schott JJ, Noireaud J, Le Marec H, Merot J, Escande D, Baro I. 14-3-3 is a regulator of the cardiac voltage-gated sodium channel Nav1.5. Circ Res. 2006;98:1538–1546. Epub 2006 May 1525. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
