

Genomic profiling of CpG methylation and allelic specificity using quantitative high-throughput mass spectrometry: critical evaluation and improvements
- PMID: 17855397
- PMCID: PMC2094090
- DOI: 10.1093/nar/gkm662
Genomic profiling of CpG methylation and allelic specificity using quantitative high-throughput mass spectrometry: critical evaluation and improvements
Abstract
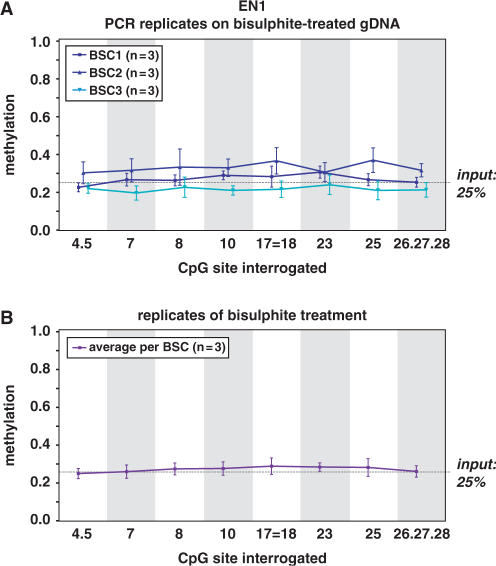
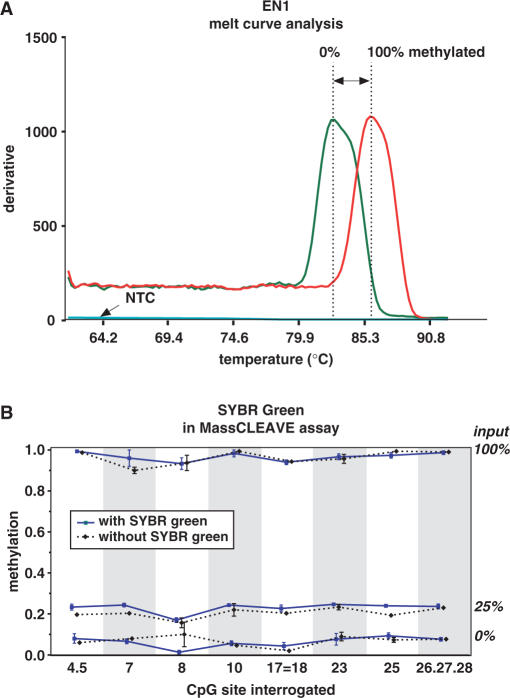
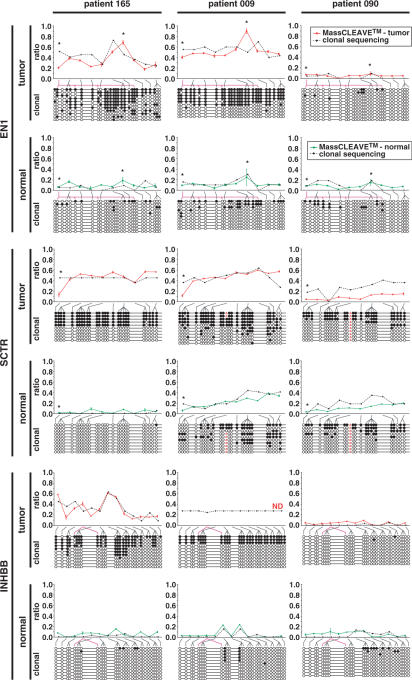
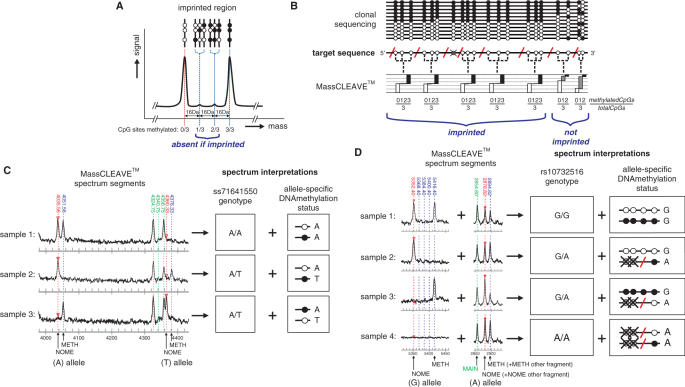
CpG methylation is a key component of the epigenome architecture that is associated with changes in gene expression without a change to the DNA sequence. Since the first reports on deregulation of DNA methylation, in diseases such as cancer, and the initiation of the Human Epigenome Project, an increasing need has arisen for a detailed, high-throughput and quantitative method of analysis to discover and validate normal and aberrant DNA methylation profiles in large sample cohorts. Here we present an improved protocol using base-specific fragmentation and MALDI-TOF mass spectrometry that enables a sensitive and high-throughput method of DNA methylation analysis, quantitative to 5% methylation for each informative CpG residue. We have determined the accuracy, variability and sensitivity of the protocol, implemented critical improvements in experimental design and interpretation of the data and developed a new formula to accurately measure CpG methylation. Key innovations now permit determination of differential and allele-specific methylation, such as in cancer and imprinting. The new protocol is ideally suitable for detailed DNA methylation analysis of multiple genomic regions and large sample cohorts that is critical for comprehensive profiling of normal and diseased human epigenomes.
Figures
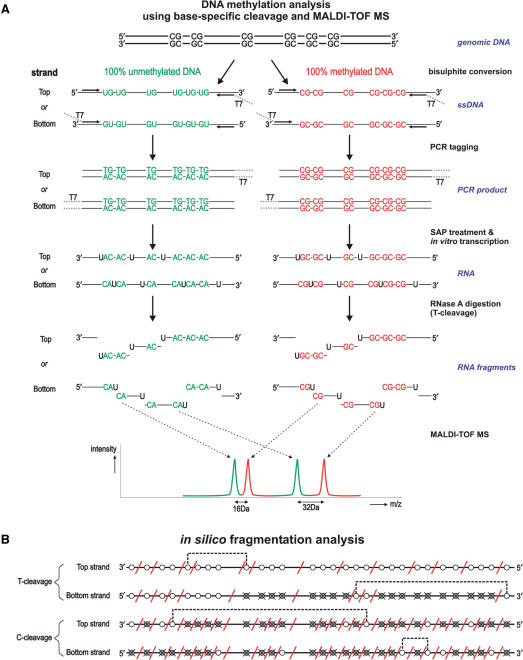
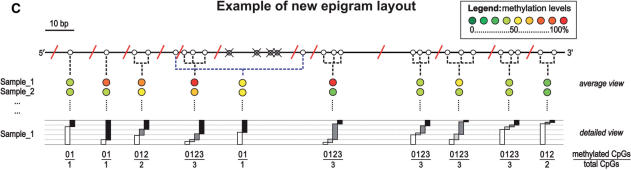
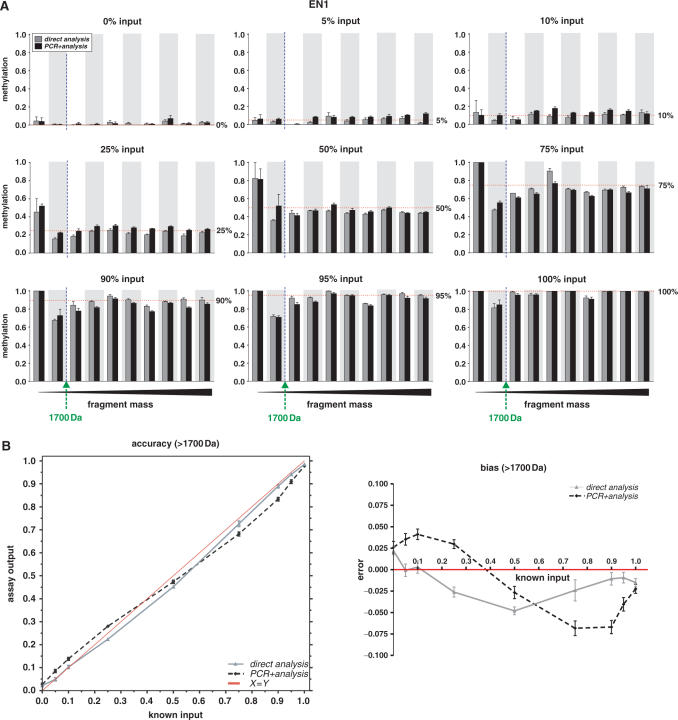
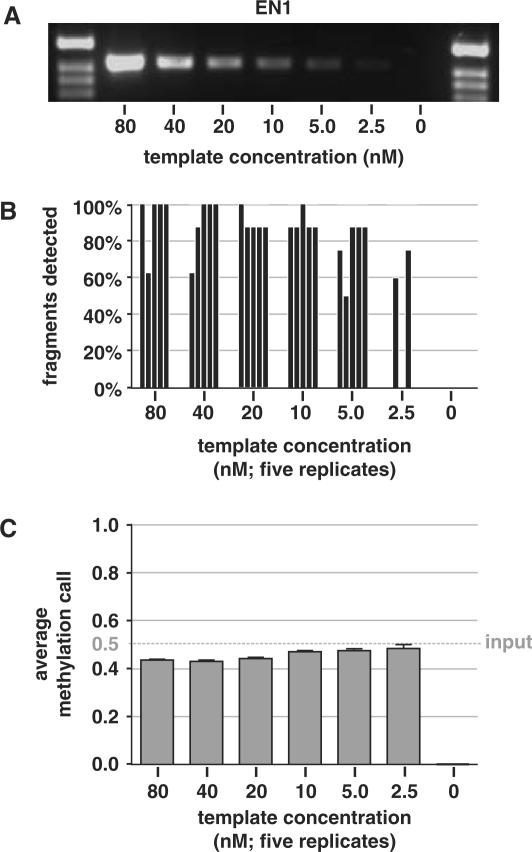
References
-
- Gardiner-Garden M, Frommer M. Transcripts and CpG islands associated with the pro-opiomelanocortin gene and other neurally expressed genes. J. Mol. Endocrinol. 1994;12:365–382. - PubMed
-
- Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat. Rev. 2006;6:107–116. - PubMed
-
- Feinberg AP, Tycko B. The history of cancer epigenetics. Nat. Rev. 2004;4:143–153. - PubMed
-
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat. Rev. 2002;3:415–428. - PubMed
-
- Morgan HD, Santos F, Green K, Dean W, Reik W. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 2005;14:R47–R58. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
