

Analysis of the role of autophagy in replication of herpes simplex virus in cell culture
- PMID: 17855538
- PMCID: PMC2169004
- DOI: 10.1128/JVI.01356-07
Analysis of the role of autophagy in replication of herpes simplex virus in cell culture
Abstract
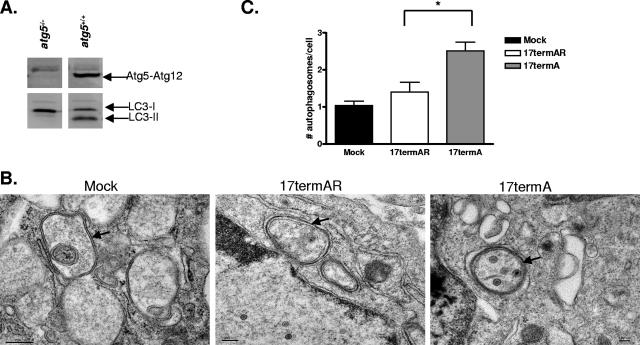
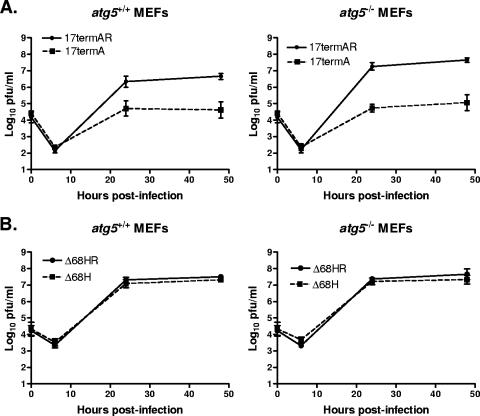
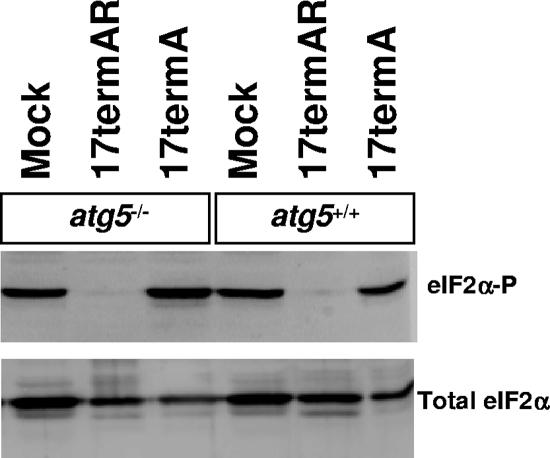
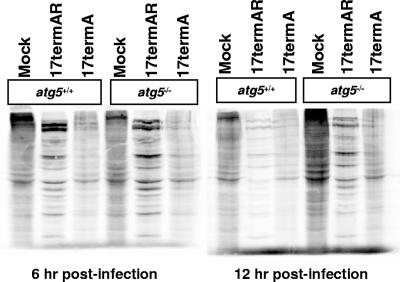
The herpes simplex virus type 1 (HSV-1) neurovirulence gene encoding ICP34.5 controls the autophagy pathway. HSV-1 strains lacking ICP34.5 are attenuated in growth and pathogenesis in animal models and in primary cultured cells. While this growth defect has been attributed to the inability of an ICP34.5-null virus to counteract the induction of translational arrest through the PKR antiviral pathway, the role of autophagy in the regulation of HSV-1 replication is unknown. Here we show that HSV-1 infection induces autophagy in primary murine embryonic fibroblasts and that autophagosome formation is increased to a greater extent following infection with an ICP34.5-deficient virus. Elimination of the autophagic pathway did not significantly alter the replication of wild-type HSV-1 or ICP34.5 mutants. The phosphorylation state of eIF2alpha and viral protein accumulation were unchanged in HSV-1-infected cells unable to undergo autophagy. These data show that while ICP34.5 regulates autophagy, it is the prevention of translational arrest by ICP34.5 rather than its control of autophagy that is the pivotal determinant of efficient HSV-1 replication in primary cell culture.
Figures
References
-
- Birmingham, C. L., A. C. Smith, M. A. Bakowski, T. Yoshimori, and J. H. Brumell. 2006. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J. Biol. Chem. 281:11374-11383. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
