

Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia
- PMID: 17873882
- PMCID: PMC2600418
- DOI: 10.1038/nm1636
Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia
Abstract
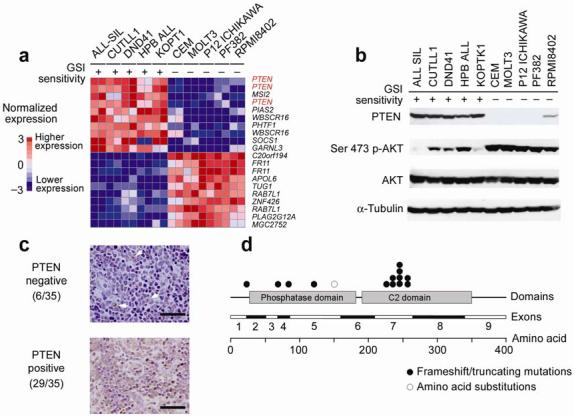
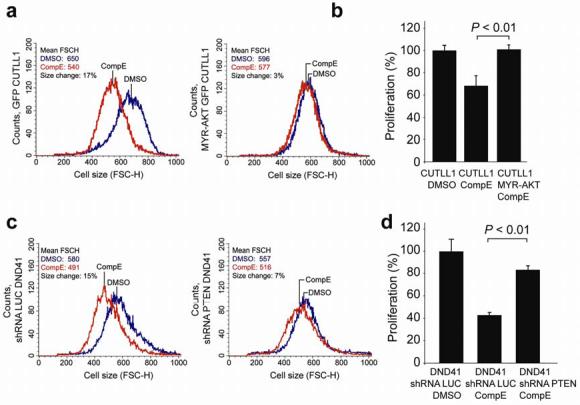
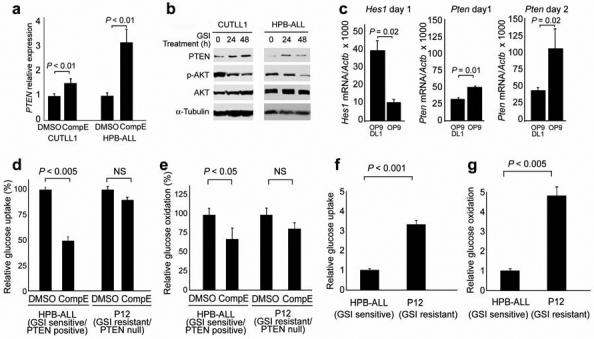
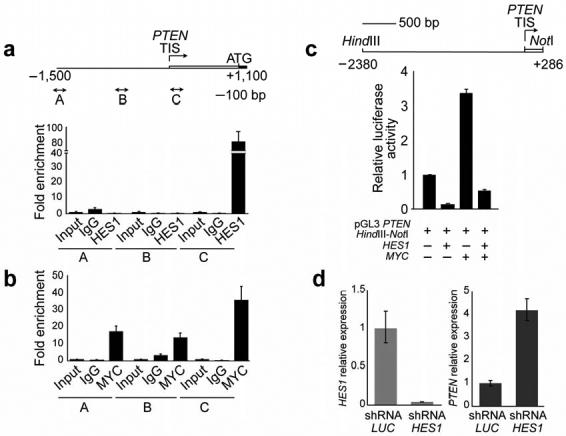
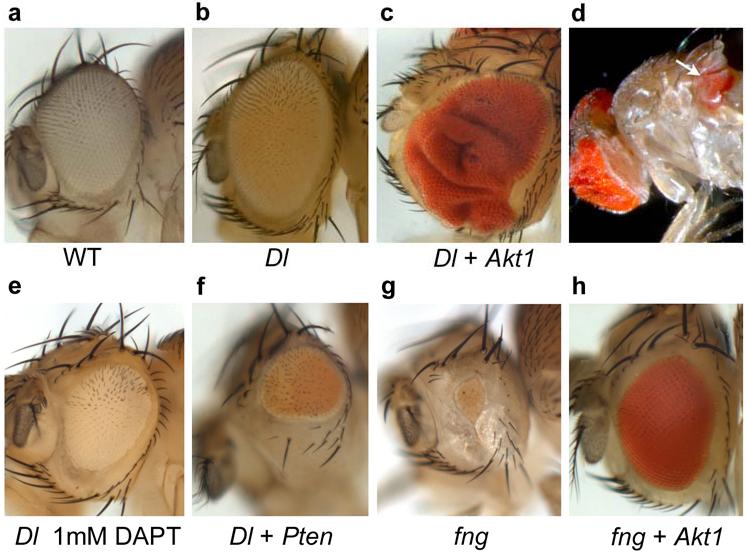
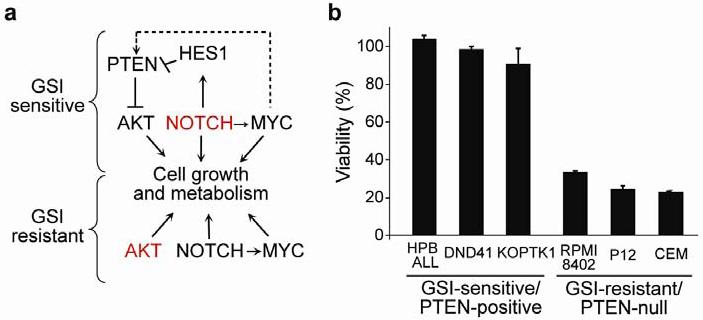
Gain-of-function mutations in NOTCH1 are common in T-cell lymphoblastic leukemias and lymphomas (T-ALL), making this receptor a promising target for drugs such as gamma-secretase inhibitors, which block a proteolytic cleavage required for NOTCH1 activation. However, the enthusiasm for these therapies has been tempered by tumor resistance and the paucity of information on the oncogenic programs regulated by oncogenic NOTCH1. Here we show that NOTCH1 regulates the expression of PTEN (encoding phosphatase and tensin homolog) and the activity of the phosphoinositol-3 kinase (PI3K)-AKT signaling pathway in normal and leukemic T cells. Notch signaling and the PI3K-AKT pathway synergize in vivo in a Drosophila melanogaster model of Notch-induced tumorigenesis, and mutational loss of PTEN is associated with human T-ALL resistance to pharmacological inhibition of NOTCH1. Overall, these findings identify transcriptional control of PTEN and regulation of the PI3K-AKT pathway as key elements of the leukemogenic program activated by NOTCH1 and provide the basis for the design of new therapeutic strategies for T-ALL.
Figures
References
-
- Grabher C, von Boehmer H, Look AT. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2006;6:347–59. - PubMed
-
- Weng AP, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–71. - PubMed
-
- Stylianou S, Clarke RB, Brennan K. Aberrant activation of notch signaling in human breast cancer. Cancer Res. 2006;66:1517–25. - PubMed
-
- Purow BW, et al. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 2005;65:2353–63. - PubMed
-
- Pahlman S, Stockhausen MT, Fredlund E, Axelson H. Notch signaling in neuroblastoma. Semin Cancer Biol. 2004;14:365–73. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
