

Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction
- PMID: 17875676
- PMCID: PMC2118469
- DOI: 10.1084/jem.20070657
Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction
Abstract
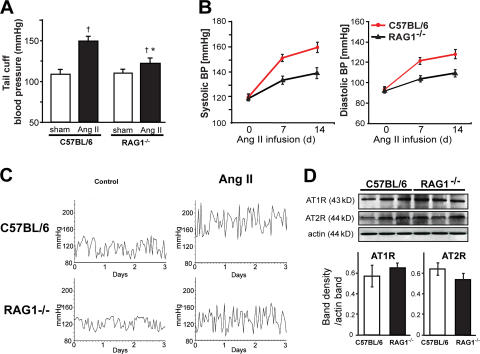
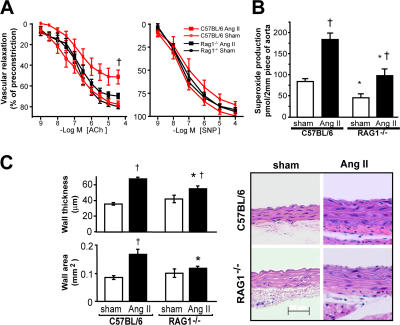
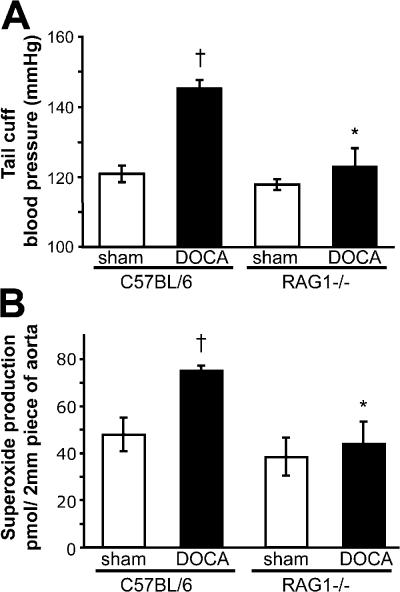
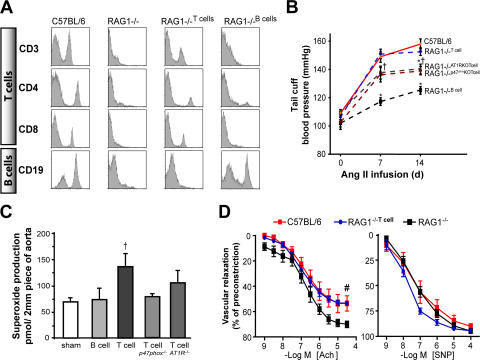
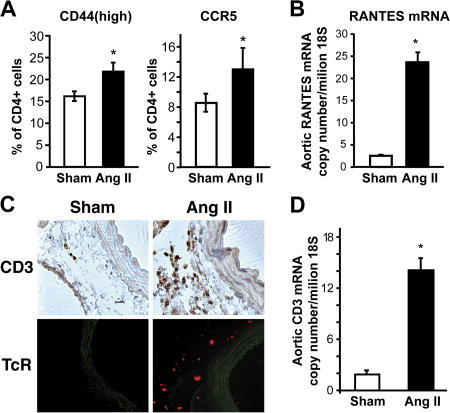
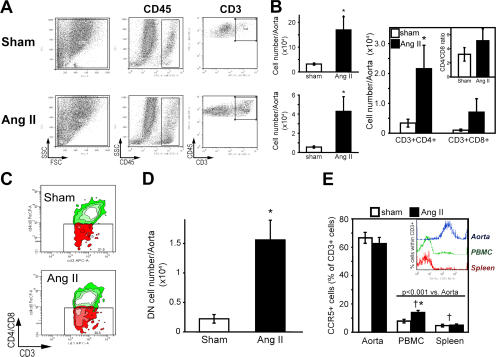
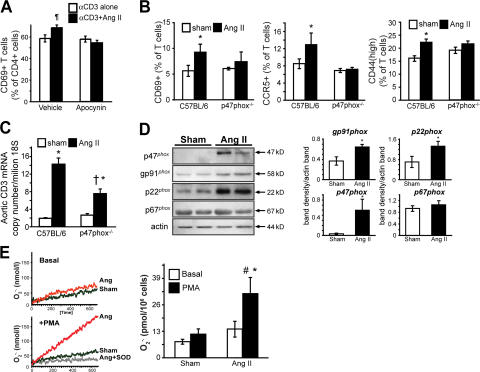
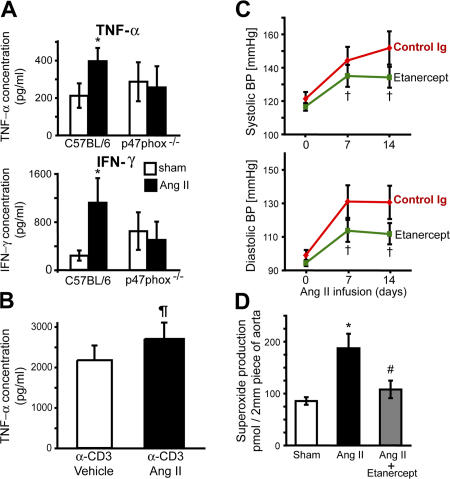
Hypertension promotes atherosclerosis and is a major source of morbidity and mortality. We show that mice lacking T and B cells (RAG-1-/- mice) have blunted hypertension and do not develop abnormalities of vascular function during angiotensin II infusion or desoxycorticosterone acetate (DOCA)-salt. Adoptive transfer of T, but not B, cells restored these abnormalities. Angiotensin II is known to stimulate reactive oxygen species production via the nicotinamide adenosine dinucleotide phosphate (NADPH) oxidase in several cells, including some immune cells. Accordingly, adoptive transfer of T cells lacking the angiotensin type I receptor or a functional NADPH oxidase resulted in blunted angiotensin II-dependent hypertension and decreased aortic superoxide production. Angiotensin II increased T cell markers of activation and tissue homing in wild-type, but not NADPH oxidase-deficient, mice. Angiotensin II markedly increased T cells in the perivascular adipose tissue (periadventitial fat) and, to a lesser extent the adventitia. These cells expressed high levels of CC chemokine receptor 5 and were commonly double negative (CD3+CD4-CD8-). This infiltration was associated with an increase in intercellular adhesion molecule-1 and RANTES in the aorta. Hypertension also increased T lymphocyte production of tumor necrosis factor (TNF) alpha, and treatment with the TNFalpha antagonist etanercept prevented the hypertension and increase in vascular superoxide caused by angiotensin II. These studies identify a previously undefined role for T cells in the genesis of hypertension and support a role of inflammation in the basis of this prevalent disease. T cells might represent a novel therapeutic target for the treatment of high blood pressure.
Figures
References
-
- Wang, Y., and Q.J. Wang. 2004. The prevalence of prehypertension and hypertension among US adults according to the new joint national committee guidelines: new challenges of the old problem. Arch. Intern. Med. 164:2126–2134. - PubMed
-
- Chobanian, A.V., G.L. Bakris, H.R. Black, W.C. Cushman, L.A. Green, J.L. Izzo Jr., D.W. Jones, B.J. Materson, S. Oparil, J.T. Wright Jr., and E.J. Roccella. 2003. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High blood pressure: the JNC 7 report. JAMA. 289:2560–2572. - PubMed
-
- Lewington, S., R. Clarke, N. Qizilbash, R. Peto, and R. Collins. 2002. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet. 360:1903–1913. - PubMed
-
- Naviar, G.L., M.C. Prieto-Carrasquero, and H. Kobori. 2006. Molecular Aspects of the renal renin-angiotensin system. In Molecular Mechanisms in Hypertension. R. Re, D. DiPette, E. Schiffrin, and R. Sowers J., editors. Taylor and Francis, London and New York. 3–14 pp.
-
- Kim, S., and H. Iwao. 2000. Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol. Rev. 52:11–34. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
