

Clinical characterization of the HOXA1 syndrome BSAS variant
- PMID: 17875913
- PMCID: PMC2826214
- DOI: 10.1212/01.wnl.0000276947.59704.cf
Clinical characterization of the HOXA1 syndrome BSAS variant
Abstract
Background: The Bosley-Salih-Alorainy syndrome (BSAS) variant of the congenital human HOXA1 syndrome results from autosomal recessive truncating HOXA1 mutations. We describe the currently recognized spectrum of ocular motility, inner ear malformations, cerebrovascular anomalies, and cognitive function.
Methods: We examined nine affected individuals from five consanguineous Saudi Arabian families, all of whom harbored the same I75-I76insG homozygous mutation in the HOXA1 gene. Patients underwent complete neurologic, neuro-ophthalmologic, orthoptic, and neuropsychological examinations. Six individuals had CT, and six had MRI of the head.
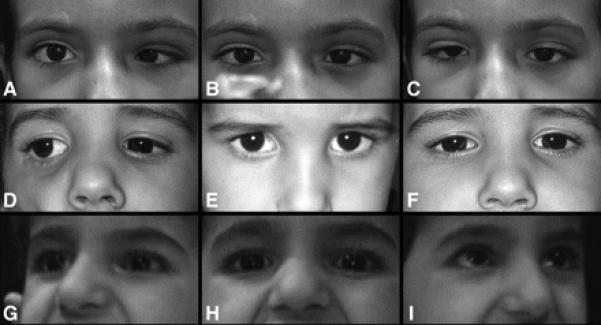
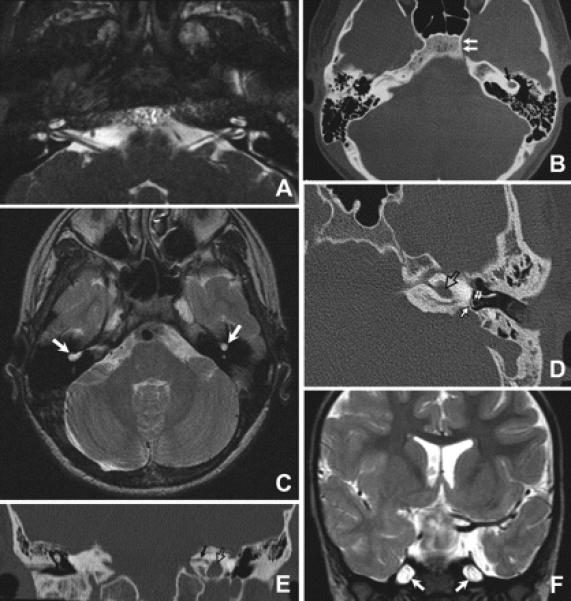
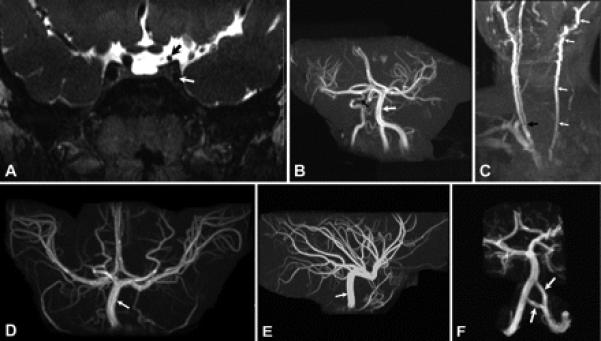
Results: All nine individuals had bilateral Duane retraction syndrome (DRS) type 3, but extent of abduction and adduction varied between eyes and individuals. Eight patients were deaf with the common cavity deformity of the inner ear, while one patient had normal hearing and skull base development. Six had delayed motor milestones, and two had cognitive and behavioral abnormalities meeting Diagnostic and Statistical Manual of Mental Disorders-IV criteria for autism spectrum disorder. MRI of the orbits, extraocular muscles, brainstem, and supratentorial brain appeared normal. All six appropriately studied patients had cerebrovascular malformations ranging from unilateral internal carotid artery hypoplasia to bilateral agenesis.
Conclusions: This report extends the Bosley-Salih-Alorainy syndrome phenotype and documents the clinical variability resulting from identical HOXA1 mutations within an isolated ethnic population. Similarities between this syndrome and thalidomide embryopathy suggest that the teratogenic effects of early thalidomide exposure in humans may be due to interaction with the HOX cascade.
Figures
References
-
- Tischfield MA, Bosley TM, Salih MA, et al. Homozygous HOXA1 mutations disrupt human brainstem, inner ear, cardiovascular and cognitive development. Nat Genet. 2005;37:1035–1037. - PubMed
-
- Chisaka O, Musci TS, Capecchi MR. Developmental defects of the ear, cranial nerves and hindbrain resulting from targeted disruption of the mouse homeobox gene Hox-1.6. Nature. 1992;355:516–520. - PubMed
-
- Lufkin T, Dierich A, LeMeur M, Mark M, Chambon P. Disruption of the Hox-1.6 homeobox gene results in defects in a region corresponding to its rostral domain of expression. Cell. 1991;66:1105–1119. - PubMed
-
- Holve S, Friedman B, Hoyme HE, et al. Athabascan brainstem dysgenesis syndrome. Am J Med Genet A. 2003;120:169–173. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases