

A framework for significance analysis of gene expression data using dimension reduction methods
- PMID: 17877799
- PMCID: PMC2194745
- DOI: 10.1186/1471-2105-8-346
A framework for significance analysis of gene expression data using dimension reduction methods
Abstract
Background: The most popular methods for significance analysis on microarray data are well suited to find genes differentially expressed across predefined categories. However, identification of features that correlate with continuous dependent variables is more difficult using these methods, and long lists of significant genes returned are not easily probed for co-regulations and dependencies. Dimension reduction methods are much used in the microarray literature for classification or for obtaining low-dimensional representations of data sets. These methods have an additional interpretation strength that is often not fully exploited when expression data are analysed. In addition, significance analysis may be performed directly on the model parameters to find genes that are important for any number of categorical or continuous responses. We introduce a general scheme for analysis of expression data that combines significance testing with the interpretative advantages of the dimension reduction methods. This approach is applicable both for explorative analysis and for classification and regression problems.
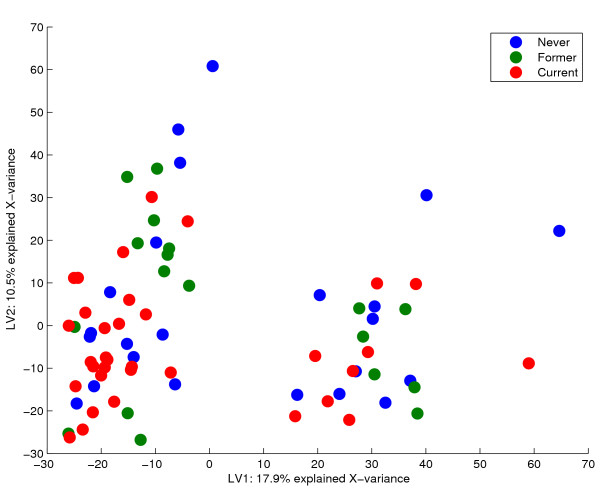
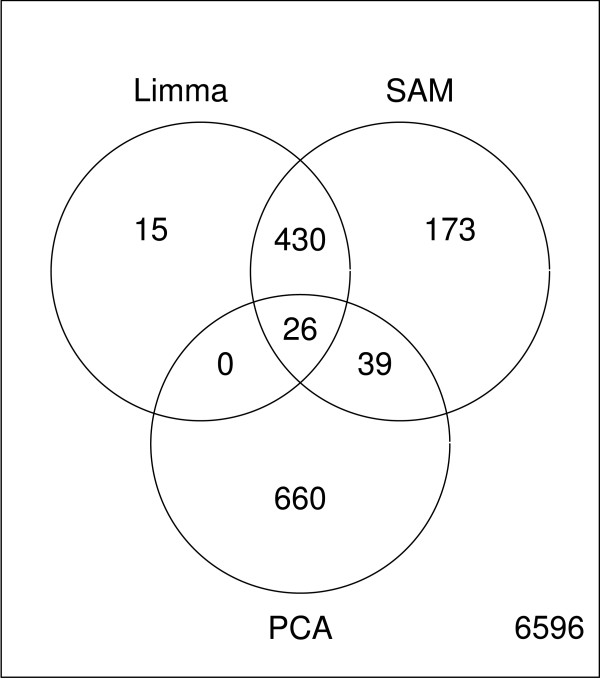
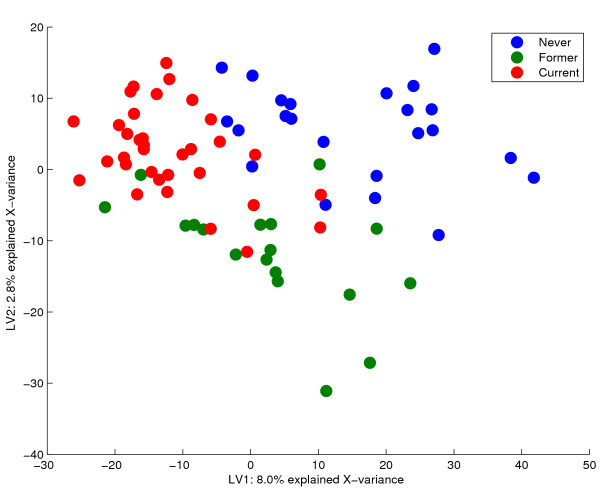
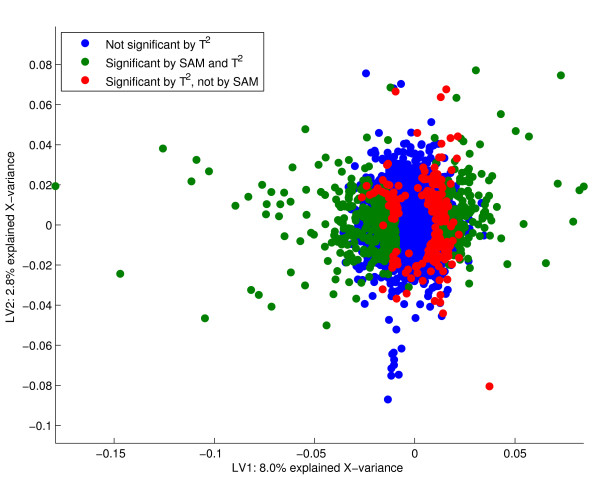
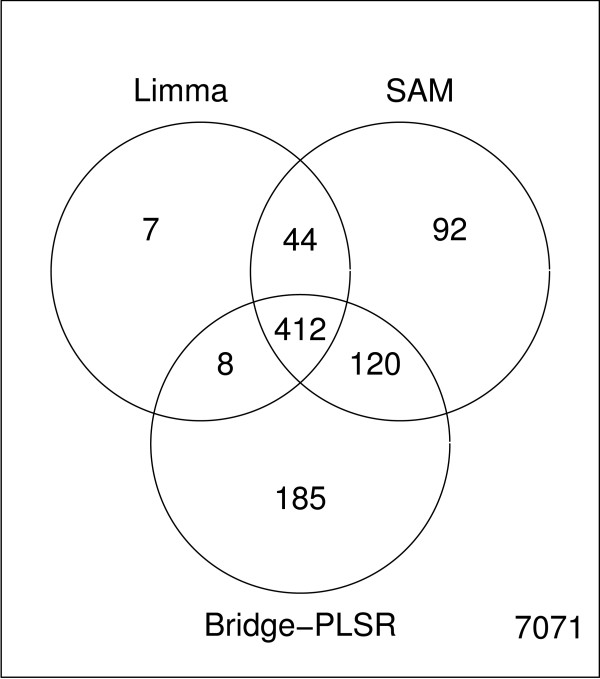
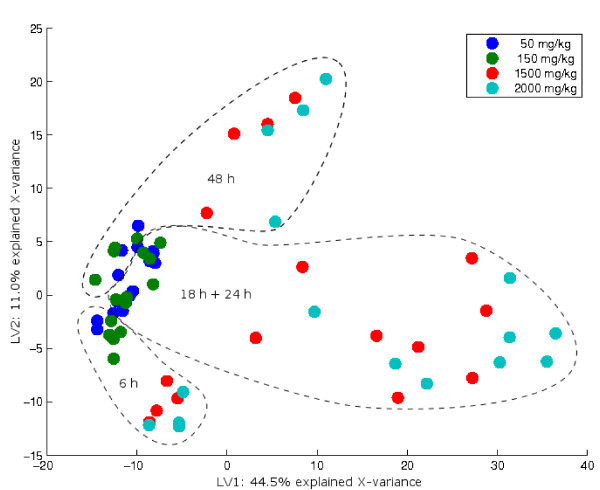
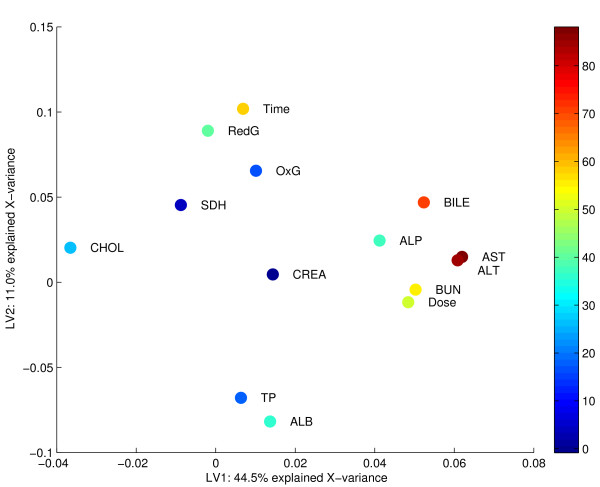
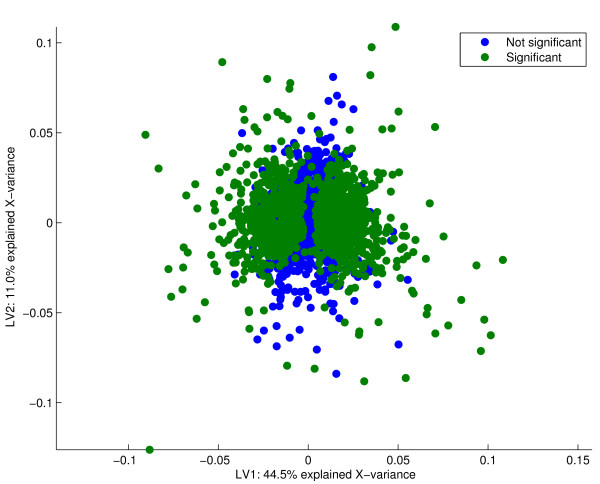
Results: Three public data sets are analysed. One is used for classification, one contains spiked-in transcripts of known concentrations, and one represents a regression problem with several measured responses. Model-based significance analysis is performed using a modified version of Hotelling's T2-test, and a false discovery rate significance level is estimated by resampling. Our results show that underlying biological phenomena and unknown relationships in the data can be detected by a simple visual interpretation of the model parameters. It is also found that measured phenotypic responses may model the expression data more accurately than if the design-parameters are used as input. For the classification data, our method finds much the same genes as the standard methods, in addition to some extra which are shown to be biologically relevant. The list of spiked-in genes is also reproduced with high accuracy.
Conclusion: The dimension reduction methods are versatile tools that may also be used for significance testing. Visual inspection of model components is useful for interpretation, and the methodology is the same whether the goal is classification, prediction of responses, feature selection or exploration of a data set. The presented framework is conceptually and algorithmically simple, and a Matlab toolbox (Mathworks Inc, USA) is supplemented.
Figures
References
-
- Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editor. Bioinformatics and computational biology solutions using R and Bioconductor. New York, USA: Springer; 2005. pp. 397–420.
-
- Martens H, Martens M. Multivariate analysis of quality: An introduction. Chichester, UK: Wiley; 2001.
-
- Wold S, Esbensen K, Geladi P. Principal component analysis. Chemometr Intell Lab. 1987;2:37–52.
-
- Boulesteix AL, Strimmer K. Partial least squares: a versatile tool for the analysis of high-dimensional genomic data. Brief Bioinformatics. 2007;8:32–44. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
