

OCPAT: an online codon-preserved alignment tool for evolutionary genomic analysis of protein coding sequences
- PMID: 17877817
- PMCID: PMC2093931
- DOI: 10.1186/1751-0473-2-5
OCPAT: an online codon-preserved alignment tool for evolutionary genomic analysis of protein coding sequences
Abstract
Background: Rapidly accumulating genome sequence data from multiple species offer powerful opportunities for the detection of DNA sequence evolution. Phylogenetic tree construction and codon-based tests for natural selection are the prevailing tools used to detect functionally important evolutionary change in protein coding sequences. These analyses often require multiple DNA sequence alignments that maintain the correct reading frame for each collection of putative orthologous sequences. Since this feature is not available in most alignment tools, codon reading frames often must be checked manually before evolutionary analyses can commence.
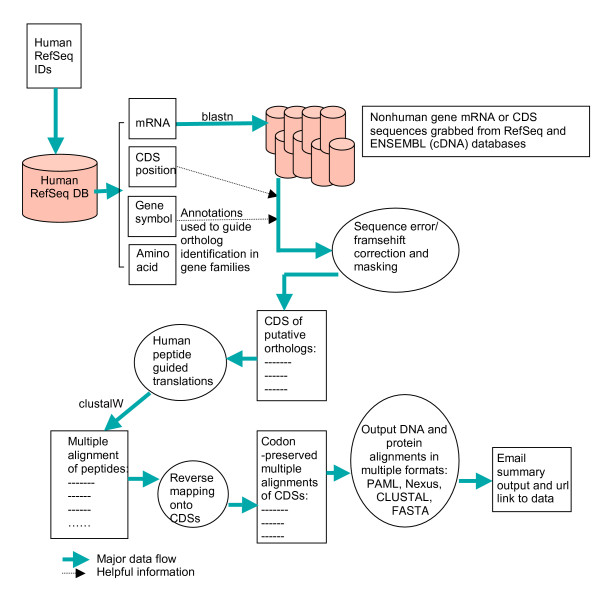
Results: Here we report an online codon-preserved alignment tool (OCPAT) that generates multiple sequence alignments automatically from the coding sequences of any list of human gene IDs and their putative orthologs from genomes of other vertebrate tetrapods. OCPAT is programmed to extract putative orthologous genes from genomes and to align the orthologs with the reading frame maintained in all species. OCPAT also optimizes the alignment by trimming the most variable alignment regions at the 5' and 3' ends of each gene. The resulting output of alignments is returned in several formats, which facilitates further molecular evolutionary analyses by appropriate available software. Alignments are generally robust and reliable, retaining the correct reading frame. The tool can serve as the first step for comparative genomic analyses of protein-coding gene sequences including phylogenetic tree reconstruction and detection of natural selection. We aligned 20,658 human RefSeq mRNAs using OCPAT. Most alignments are missing sequence(s) from at least one species; however, functional annotation clustering of the ~1700 transcripts that were alignable to all species shows that genes involved in multi-subunit protein complexes are highly conserved.
Conclusion: The OCPAT program facilitates large-scale evolutionary and phylogenetic analyses of entire biological processes, pathways, and diseases.
Figures
Similar articles
-
[Analysis, identification and correction of some errors of model refseqs appeared in NCBI Human Gene Database by in silico cloning and experimental verification of novel human genes].Yi Chuan Xue Bao. 2004 May;31(5):431-43. Yi Chuan Xue Bao. 2004. PMID: 15478601 Chinese.
-
transAlign: using amino acids to facilitate the multiple alignment of protein-coding DNA sequences.BMC Bioinformatics. 2005 Jun 22;6:156. doi: 10.1186/1471-2105-6-156. BMC Bioinformatics. 2005. PMID: 15969769 Free PMC article.
-
CGAT: a comparative genome analysis tool for visualizing alignments in the analysis of complex evolutionary changes between closely related genomes.BMC Bioinformatics. 2006 Oct 24;7:472. doi: 10.1186/1471-2105-7-472. BMC Bioinformatics. 2006. PMID: 17062155 Free PMC article.
-
BIR Pipeline for Preparation of Phylogenomic Data.Evol Bioinform Online. 2015 Apr 27;11:79-83. doi: 10.4137/EBO.S10189. eCollection 2015. Evol Bioinform Online. 2015. PMID: 25987827 Free PMC article. Review.
-
Computation and analysis of genomic multi-sequence alignments.Annu Rev Genomics Hum Genet. 2007;8:193-213. doi: 10.1146/annurev.genom.8.080706.092300. Annu Rev Genomics Hum Genet. 2007. PMID: 17489682 Review.
Cited by
-
Phylogenomic analyses reveal convergent patterns of adaptive evolution in elephant and human ancestries.Proc Natl Acad Sci U S A. 2009 Dec 8;106(49):20824-9. doi: 10.1073/pnas.0911239106. Epub 2009 Nov 19. Proc Natl Acad Sci U S A. 2009. PMID: 19926857 Free PMC article.
-
Distinct genomic signatures of adaptation in pre- and postnatal environments during human evolution.Proc Natl Acad Sci U S A. 2008 Mar 4;105(9):3215-20. doi: 10.1073/pnas.0712400105. Epub 2008 Feb 27. Proc Natl Acad Sci U S A. 2008. PMID: 18305157 Free PMC article.
-
Inhibitory interneurons of the human prefrontal cortex display conserved evolution of the phenotype and related genes.Proc Biol Sci. 2010 Apr 7;277(1684):1011-20. doi: 10.1098/rspb.2009.1831. Epub 2009 Dec 2. Proc Biol Sci. 2010. PMID: 19955152 Free PMC article.
-
Development and evaluation of new mask protocols for gene expression profiling in humans and chimpanzees.BMC Bioinformatics. 2009 Mar 5;10:77. doi: 10.1186/1471-2105-10-77. BMC Bioinformatics. 2009. PMID: 19265541 Free PMC article.
-
Phylogeny of the Ferungulata (Mammalia: Laurasiatheria) as determined from phylogenomic data.Mol Phylogenet Evol. 2009 Sep;52(3):660-4. doi: 10.1016/j.ympev.2009.05.002. Epub 2009 May 10. Mol Phylogenet Evol. 2009. PMID: 19435603 Free PMC article.
References
-
- Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. - PubMed
LinkOut - more resources
Full Text Sources
