

Cholesterol suppresses cellular TGF-beta responsiveness: implications in atherogenesis
- PMID: 17878231
- PMCID: PMC2045688
- DOI: 10.1242/jcs.006916
Cholesterol suppresses cellular TGF-beta responsiveness: implications in atherogenesis
Abstract
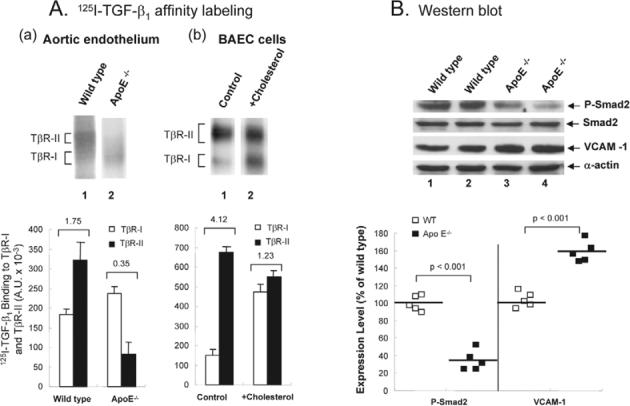
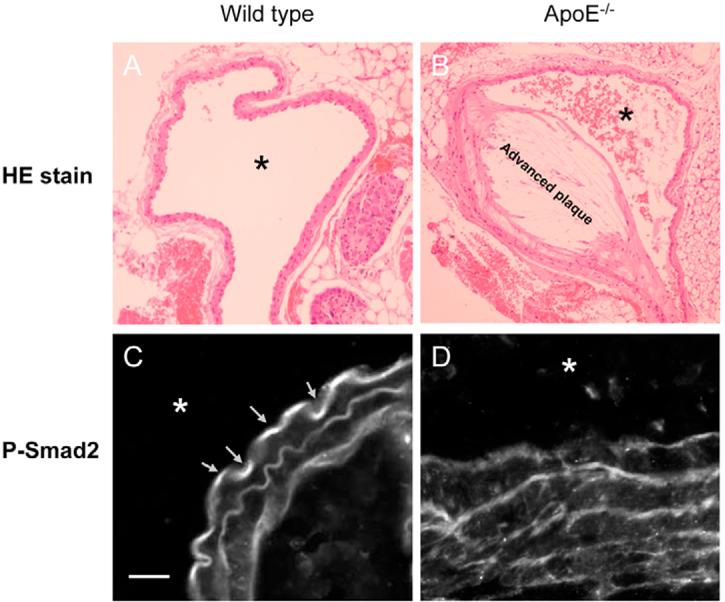
Hypercholesterolemia is a major causative factor for atherosclerotic cardiovascular disease. The molecular mechanisms by which cholesterol initiates and facilitates the process of atherosclerosis are not well understood. Here, we demonstrate that cholesterol treatment suppresses or attenuates TGF-beta responsiveness in all cell types studied as determined by measuring TGF-beta-induced Smad2 phosphorylation and nuclear translocation, TGF-beta-induced PAI-1 expression, TGF-beta-induced luciferase reporter gene expression and TGF-beta-induced growth inhibition. Cholesterol, alone or complexed in lipoproteins (LDL, VLDL), suppresses TGF-beta responsiveness by increasing lipid raft and/or caveolae accumulation of TGF-beta receptors and facilitating rapid degradation of TGF-beta and thus suppressing TGF-beta-induced signaling. Conversely, cholesterol-lowering agents (fluvastatin and lovastatin) and cholesterol-depleting agents (beta-cyclodextrin and nystatin) enhance TGF-beta responsiveness by increasing non-lipid raft microdomain accumulation of TGF-beta receptors and facilitating TGF-beta-induced signaling. Furthermore, the effects of cholesterol on the cultured cells are also found in the aortic endothelium of ApoE-null mice fed a high-cholesterol diet. These results suggest that high cholesterol contributes to atherogenesis, at least in part, by suppressing TGF-beta responsiveness in vascular cells.
Figures








References
-
- Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, Rifkin DB. An essay for transforming growth factor-β using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal. Biochem. 1994;216:276–284. - PubMed
-
- Alberts AW. Discovery, biochemistry and biology of lovastatin. Am. J. Cardiol. 1988;62:11. - PubMed
-
- Bickel PE. Lipid rafts and insulin signaling. Am. J. Physiol. Endocrinol. Metab. 2002;282:E1–E10. - PubMed
-
- Brown MS, Goldstein JL. Lowering LDL – not only how low, but how long? Science. 2006;311:1721–1723. - PubMed
-
- Buchwald H. Cholesterol inhibition, cancer, and chemotherapy. Lancet. 1992;339:1154–1156. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
