

Global control of aberrant splice-site activation by auxiliary splicing sequences: evidence for a gradient in exon and intron definition
- PMID: 17881373
- PMCID: PMC2095810
- DOI: 10.1093/nar/gkm680
Global control of aberrant splice-site activation by auxiliary splicing sequences: evidence for a gradient in exon and intron definition
Abstract
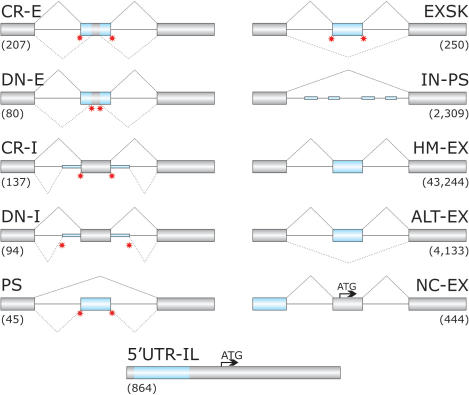
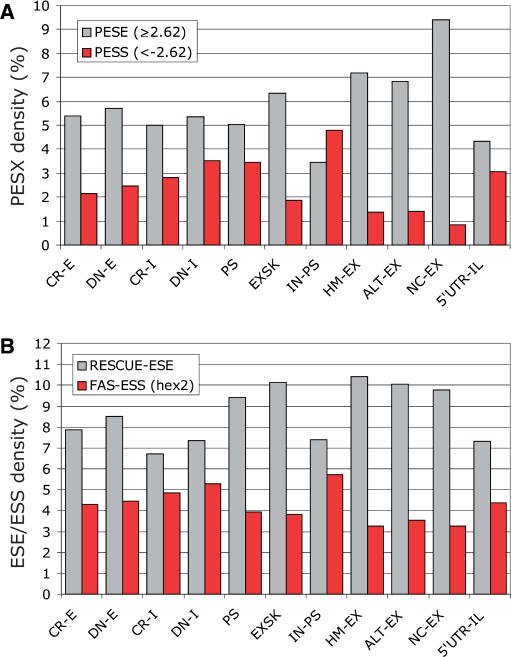
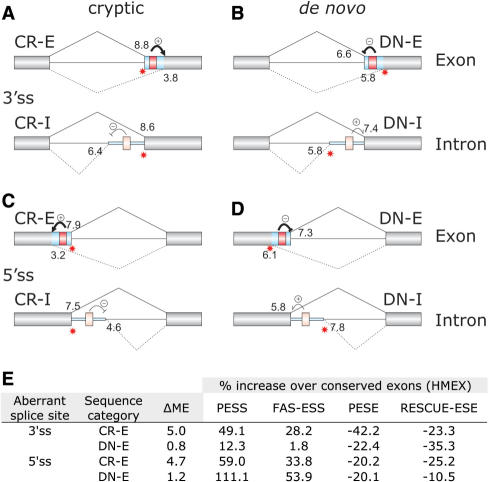
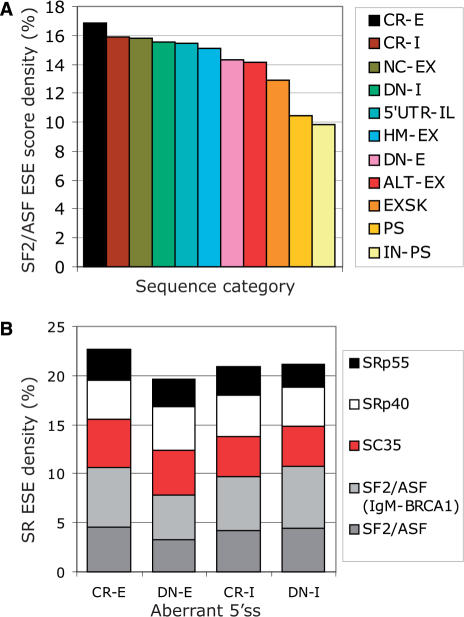
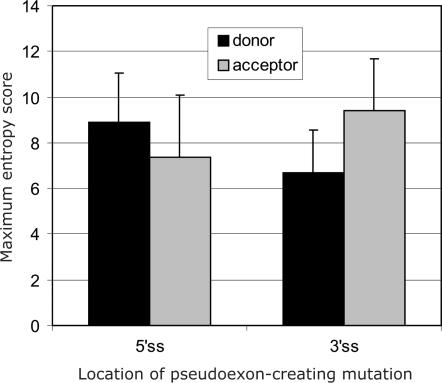
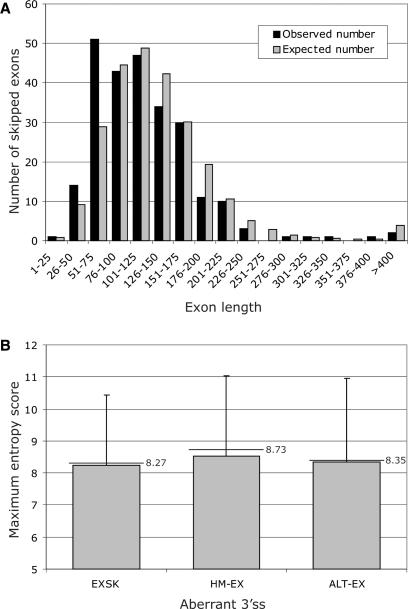
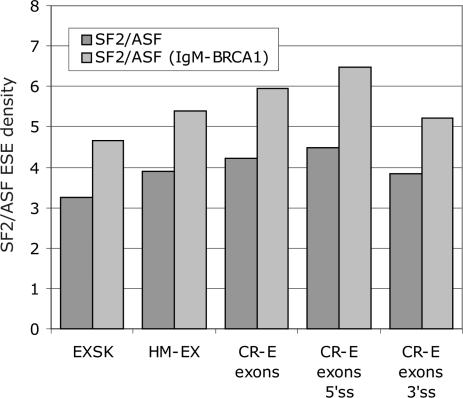
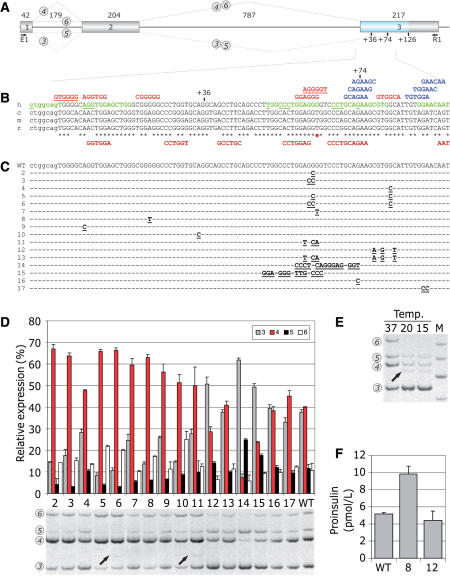
Auxiliary splicing signals play a major role in the regulation of constitutive and alternative pre-mRNA splicing, but their relative importance in selection of mutation-induced cryptic or de novo splice sites is poorly understood. Here, we show that exonic sequences between authentic and aberrant splice sites that were activated by splice-site mutations in human disease genes have lower frequencies of splicing enhancers and higher frequencies of splicing silencers than average exons. Conversely, sequences between authentic and intronic aberrant splice sites have more enhancers and less silencers than average introns. Exons that were skipped as a result of splice-site mutations were smaller, had lower SF2/ASF motif scores, a decreased availability of decoy splice sites and a higher density of silencers than exons in which splice-site mutation activated cryptic splice sites. These four variables were the strongest predictors of the two aberrant splicing events in a logistic regression model. Elimination or weakening of predicted silencers in two reporters consistently promoted use of intron-proximal splice sites if these elements were maintained at their original positions, with their modular combinations producing expected modification of splicing. Together, these results show the existence of a gradient in exon and intron definition at the level of pre-mRNA splicing and provide a basis for the development of computational tools that predict aberrant splicing outcomes.
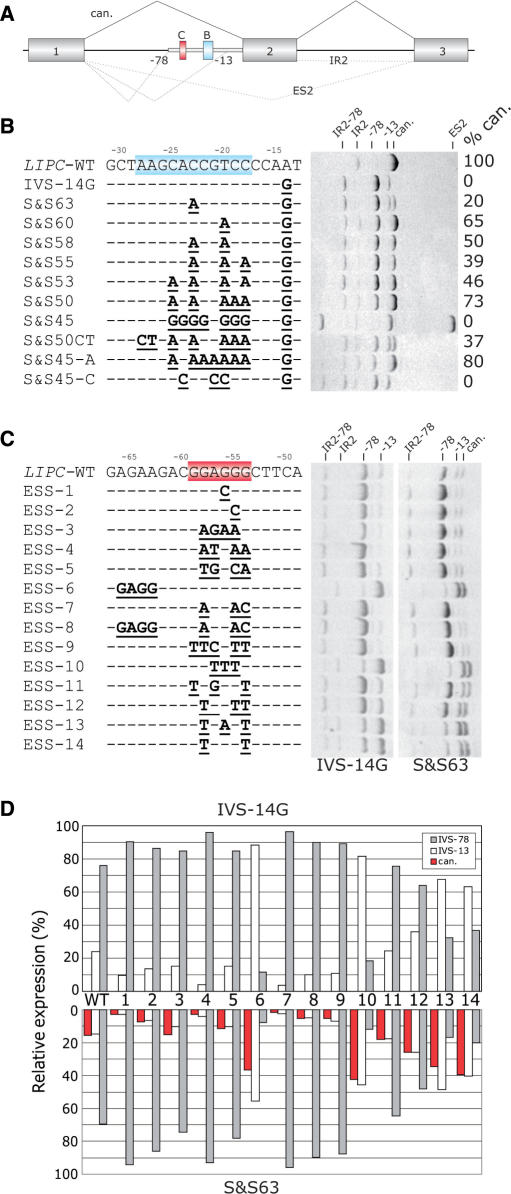
Figures
Similar articles
-
Computational analysis of splicing errors and mutations in human transcripts.BMC Genomics. 2008 Jan 14;9:13. doi: 10.1186/1471-2164-9-13. BMC Genomics. 2008. PMID: 18194514 Free PMC article.
-
Extensive in silico analysis of NF1 splicing defects uncovers determinants for splicing outcome upon 5' splice-site disruption.Hum Mutat. 2007 Jun;28(6):599-612. doi: 10.1002/humu.20493. Hum Mutat. 2007. PMID: 17311297
-
Biased exon/intron distribution of cryptic and de novo 3' splice sites.Nucleic Acids Res. 2005 Sep 1;33(15):4882-98. doi: 10.1093/nar/gki811. Print 2005. Nucleic Acids Res. 2005. PMID: 16141195 Free PMC article.
-
Alterations of pre-mRNA splicing in cancer.Genes Chromosomes Cancer. 2005 Apr;42(4):342-57. doi: 10.1002/gcc.20156. Genes Chromosomes Cancer. 2005. PMID: 15648050 Review.
-
Unannotated splicing regulatory elements in deep intron space.Wiley Interdiscip Rev RNA. 2021 Sep;12(5):e1656. doi: 10.1002/wrna.1656. Epub 2021 Apr 22. Wiley Interdiscip Rev RNA. 2021. PMID: 33887804 Review.
Cited by
-
A Dual Reporter Splicing Assay Using HaloTag-containing Proteins.Curr Chem Genomics. 2012;6:27-37. doi: 10.2174/1875397301206010027. Epub 2012 Sep 20. Curr Chem Genomics. 2012. PMID: 23136623 Free PMC article.
-
Exonic splicing code and coordination of divalent metals in proteins.Nucleic Acids Res. 2024 Feb 9;52(3):1090-1106. doi: 10.1093/nar/gkad1161. Nucleic Acids Res. 2024. PMID: 38055834 Free PMC article.
-
Dual effect of a single nucleotide polymorphism in the first intron of the porcine secreted phosphoprotein 1 gene: allele-specific binding of C/EBP beta and activation of aberrant splicing.BMC Mol Biol. 2009 Oct 21;10:96. doi: 10.1186/1471-2199-10-96. BMC Mol Biol. 2009. PMID: 19845957 Free PMC article.
-
Characterization of NOL7 gene point mutations, promoter methylation, and protein expression in cervical cancer.Int J Gynecol Pathol. 2012 Jan;31(1):15-24. doi: 10.1097/PGP.0b013e318220ba16. Int J Gynecol Pathol. 2012. PMID: 22123719 Free PMC article.
-
Variation of Genes Encoding Tryptophan Catabolites Pathway Enzymes in Stroke.J Clin Med. 2019 Dec 3;8(12):2133. doi: 10.3390/jcm8122133. J Clin Med. 2019. PMID: 31817010 Free PMC article.
References
-
- Caputi M, Zahler AM. Determination of the RNA binding specificity of the heterogeneous nuclear ribonucleoprotein (hnRNP) H/H'/F/2H9 family. J. Biol. Chem. 2001;276:43850–43859. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
