

Sodium channel Scn1b null mice exhibit prolonged QT and RR intervals
- PMID: 17884088
- PMCID: PMC2099572
- DOI: 10.1016/j.yjmcc.2007.07.062
Sodium channel Scn1b null mice exhibit prolonged QT and RR intervals
Abstract
In neurons, voltage-gated sodium channel beta subunits regulate the expression levels, subcellular localization, and electrophysiological properties of sodium channel alpha subunits. However, the contribution of beta subunits to sodium channel function in heart is poorly understood. We examined the role of beta1 in cardiac excitability using Scn1b null mice. Compared to wildtype mice, electrocardiograms recorded from Scn1b null mice displayed longer RR intervals and extended QT(c) intervals, both before and after autonomic block. In acutely dissociated ventricular myocytes, loss of beta1 expression resulted in a approximately 1.6-fold increase in both peak and persistent sodium current while channel gating and kinetics were unaffected. Na(v)1.5 expression increased in null myocytes approximately 1.3-fold. Action potential recordings in acutely dissociated ventricular myocytes showed slowed repolarization, supporting the extended QT(c) interval. Immunostaining of individual myocytes or ventricular sections revealed no discernable alterations in the localization of sodium channel alpha or beta subunits, ankyrin(B), ankyrin(G), N-cadherin, or connexin-43. Together, these results suggest that beta1 is critical for normal cardiac excitability and loss of beta1 may be associated with a long QT phenotype.
Figures
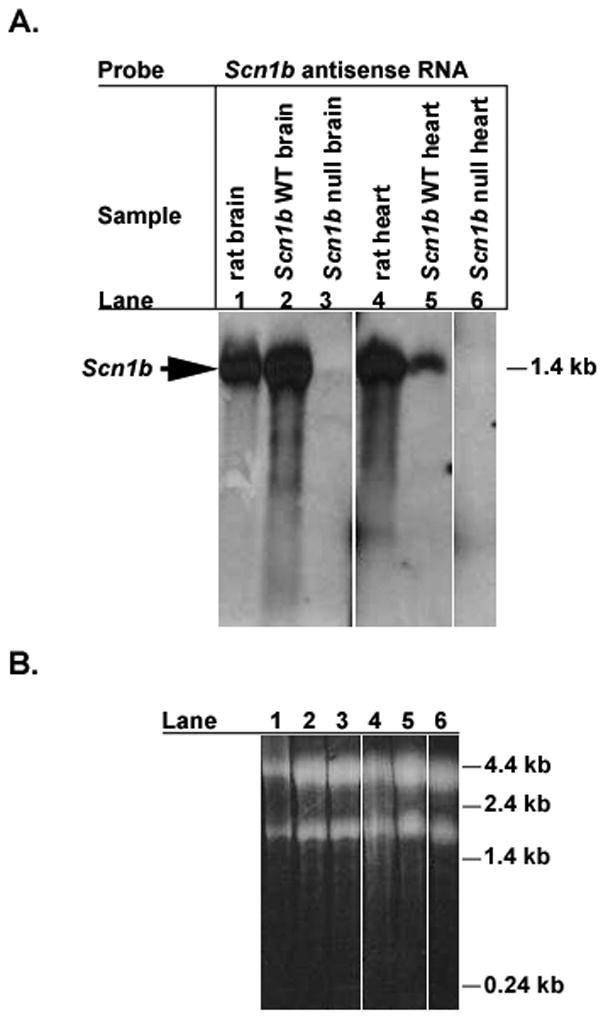
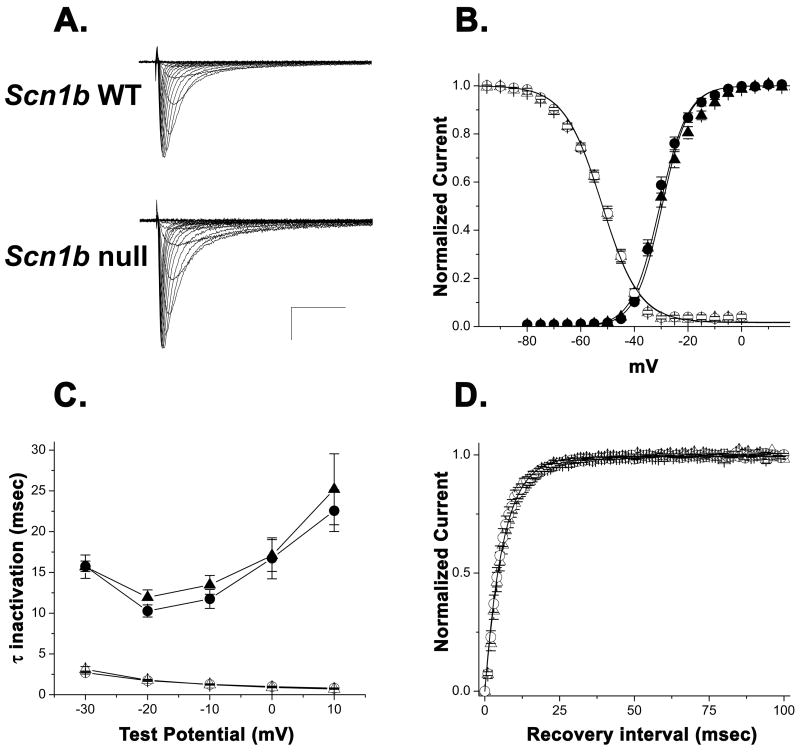

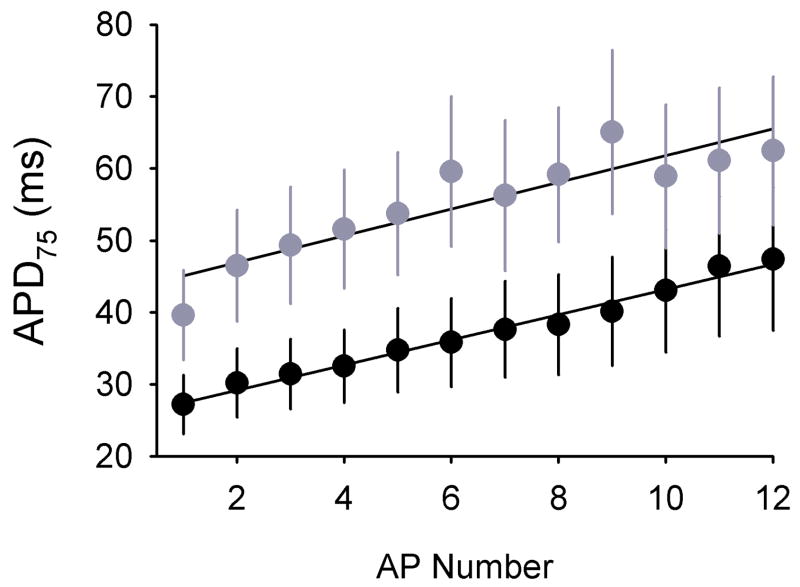



References
-
- Tan HL, Bezzina CR, Smits JP, Verkerk AO, Wilde AA. Genetic control of sodium channel function. Cardiovasc Res. 2003 Mar 15;57(4):961–73. - PubMed
-
- Meadows LS, Isom LL. Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovascular Research. 2005;67:448–58. - PubMed
-
- Hartshorne RP, Messner DJ, Coppersmith JC, Catterall WA. The saxitoxin receptor of the sodium channel from rat brain Evidence for two nonidentical beta subunits. JBiolChem. 1982;257:13888–91. - PubMed
-
- Isom LL, De Jongh KS, Patton DE, Reber BFX, Offord J, Charbonneau H, et al. Primary structure and functional expression of the β1 subunit of the rat brain sodium channel. Science. 1992;256:839–42. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
