

TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor-1
- PMID: 17884256
- PMCID: PMC3191465
- DOI: 10.1016/j.neuint.2007.08.006
TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor-1
Abstract
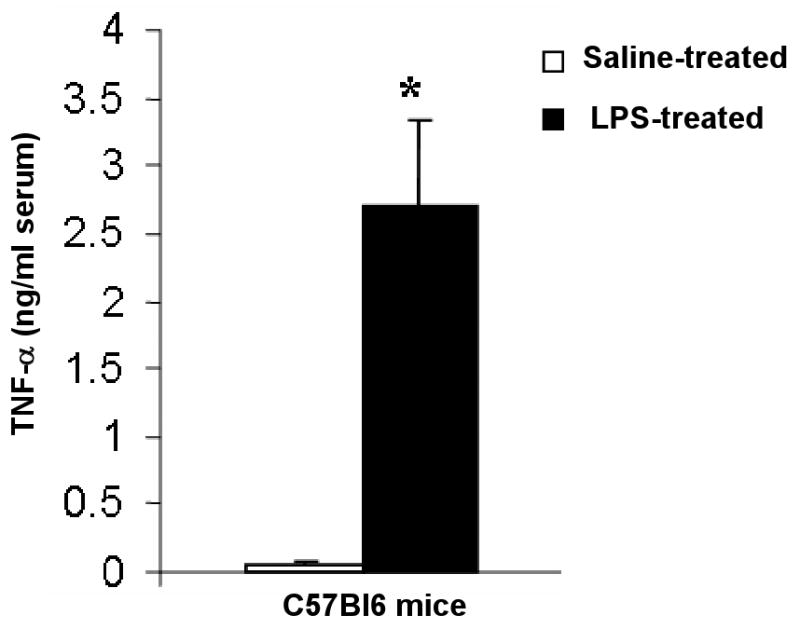
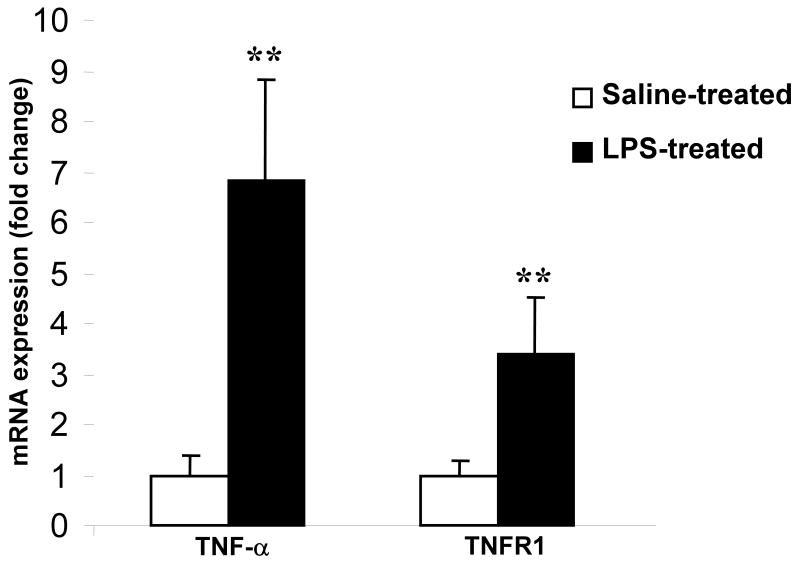
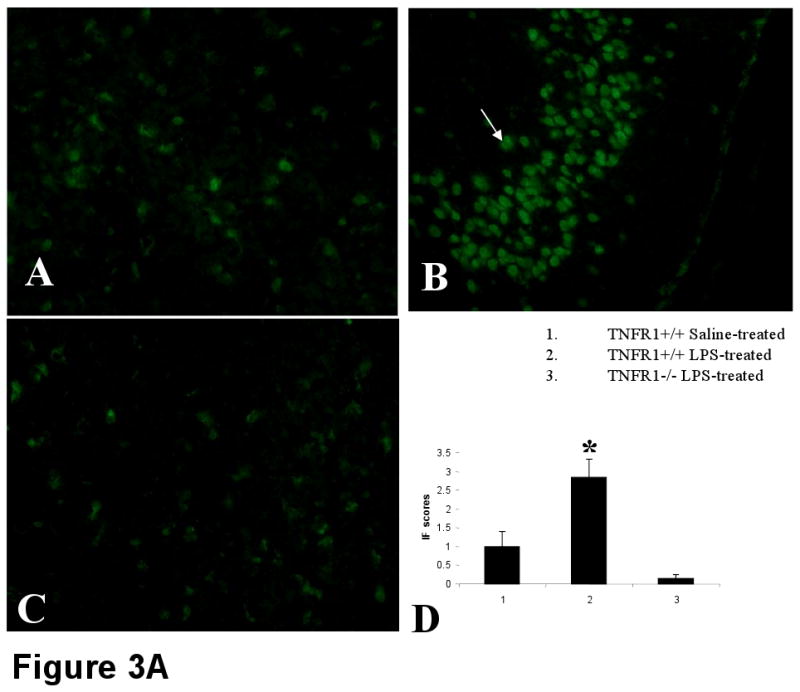
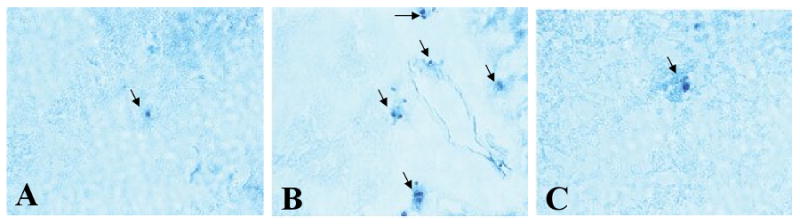
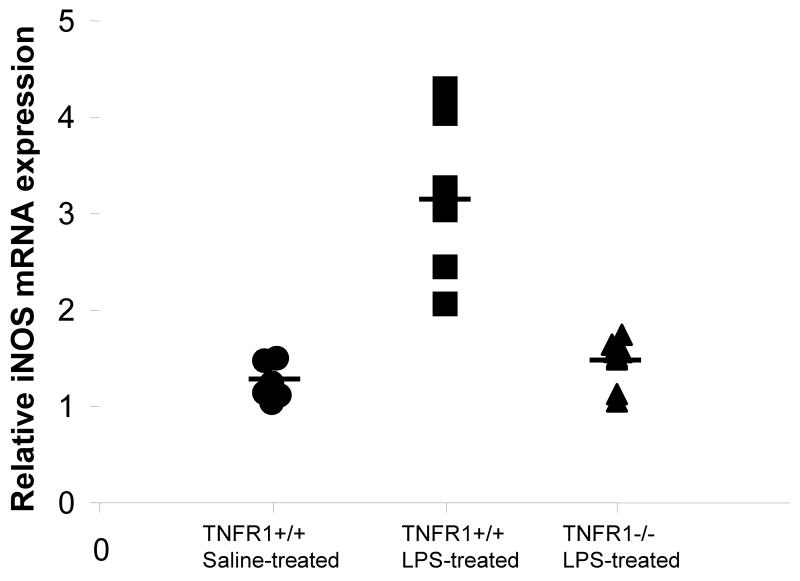
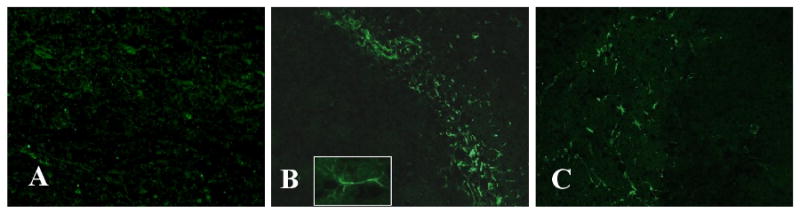
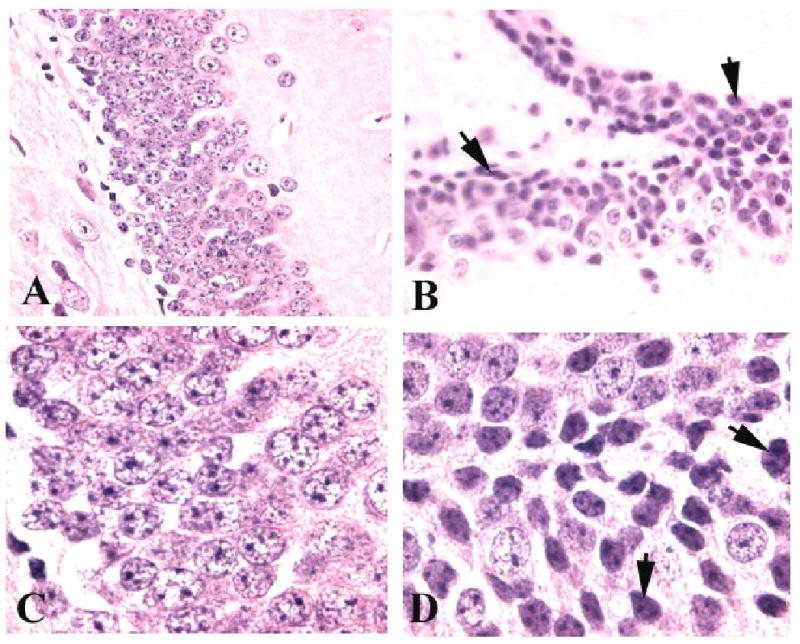
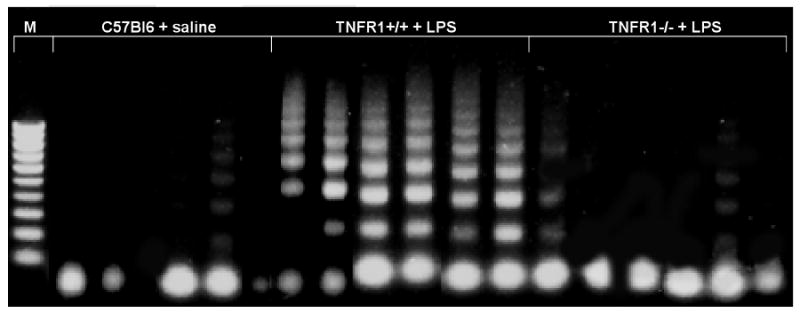
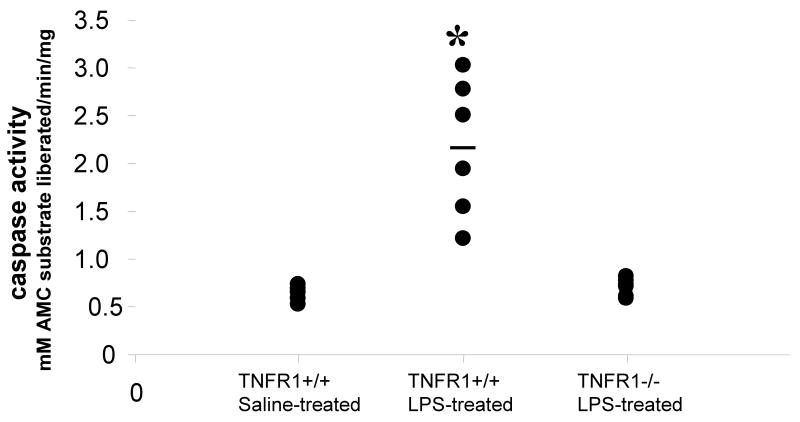
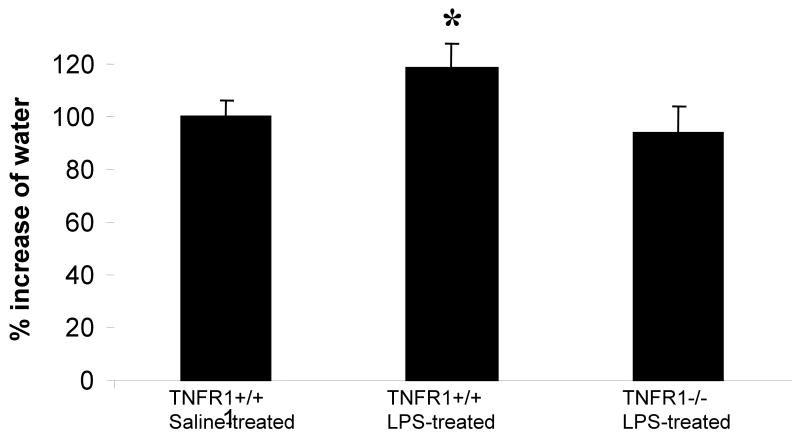
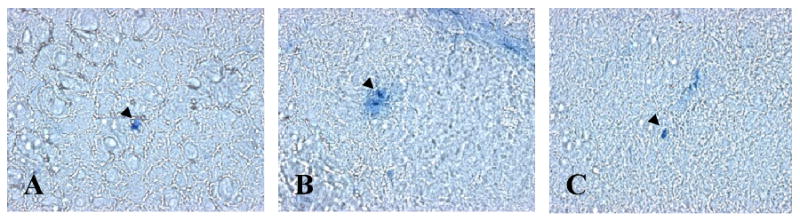
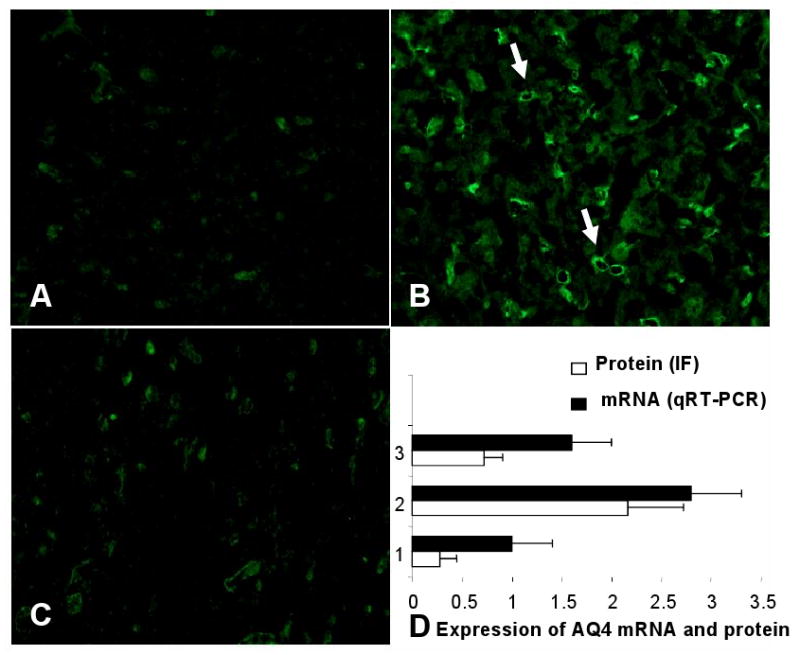
In this study, we demonstrate that mice deficient in TNFR1 (TNFR1(-/-)) were resistant to LPS-induced encephalopathy. Systemic administration of lipopolysaccharide (LPS) induces a widespread inflammatory response similar to that observed in sepsis. Following LPS administration TNFR1(-/-) mice had less caspase-dependent apoptosis in brain cells and fewer neutrophils infiltrating the brain (p<0.039), compared to control C57Bl6 (TNFR1(+/+)) mice. TNFR1-dependent increase in aquaporin (AQP)-4 mRNA and protein expression was observed with a concomitant increase in water content, in brain (18% increase in C57Bl6 mice treated with LPS versus those treated with saline), similar to cerebral edema observed in sepsis. Furthermore, absence of TNFR1 partially but significantly reduced the activation of astrocytes, as shown by immunofluorescence and markedly inhibited iNOS mRNA expression (p<0.01). Septic encephalopathy is a devastating complication of sepsis. Although, considerable work has been done to identify the mechanism causing the pathological alterations in this setting, the culprit still remains an enigma. Our results demonstrate for the first time that endotoxemia leads to inflammation in brain, with alteration in blood-brain barrier, up-regulation of AQP4 and associated edema, neutrophil infiltration, astrocytosis, as well as apoptotic cellular death, all of which appear to be mediated by TNF-alpha signaling through TNFR1.
Figures
References
-
- Alexander JJ, Bao L, Jacob A, Kraus DM, Holers VM, Quigg RJ. Administration of the soluble complement inhibitor, Crry-Ig, reduces inflammation and aquaporin 4 expression in lupus cerebritis. Biochim Biophys Acta. 2003;1639:169–176. - PubMed
-
- Alexander JJ, Jacob A, Bao L, MacDonald RL, Quigg RJ. Complement-Dependent Apoptosis and Inflammatory Gene Changes in Murine Lupus Cerebritis. J Immunol. 2005;175:8312–8319. - PubMed
-
- Alexander JJ, Jacob A, Vezina P, Sekine H, Gilkeson GS, Quigg RJ. Absence of functional alternative complement pathway alleviates lupus cerebritis. Eur J Immunol. 2007;37:1691–1701. - PubMed
-
- Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2:734–744. - PubMed
-
- Amrani Y, Lazaar AL, Hoffman R, Amin K, Ousmer S, Panettieri RA., Jr Activation of p55 tumor necrosis factor-alpha receptor-1 coupled to tumor necrosis factor receptor-associated factor 2 stimulates intercellular adhesion molecule-1 expression by modulating a thapsigargin-sensitive pathway in human tracheal smooth muscle cells. Mol Pharmacol. 2000;58:237–245. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
